Enteric-type adenocarcinoma of the lung harbouring a novel KRAS Q22K mutation with concomitant KRAS polysomy: a case report
Giulio Metro1, Emanuele Valtorta2, Annamaria Siggillino1, Calogero Lauricella2, Matteo Cenci1, Vienna Ludovini1, Elisa Minenza3, Enrico Prosperi4, Biagio Ricciuti1, Alberto Rebonato5, Alessandra Bassetti6 and Lucio Crinò1
1 Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia 06156, Italy
2 Niguarda Cancer Center, Division of Pathology, Ospedale Niguarda Ca’ Granda, Milano 20162, Italy
3 Medical Oncology, Santa Maria Hospital, Azienda Ospedaliera di Terni, Terni 05100, Italy
4 Department of Experimental Medicine, Pathological Anatomy and Histology Unit, School of Medicine, University of Perugia, Perugia 06156, Italy
5 Department of Diagnostic Imaging, Santa Maria della Misericordia Hospital, University of Perugia 06156, Italy
6 Medical Oncology, Narni Hospital, Narni (TR) 05035, Italy
Correspondence to: Giulio Metro, Email: giulio.metro@yahoo.com
Abstract
This case describes a novel KRAS Q22K mutation with simultaneous KRAS polysomy in a patient with advanced, enteric-type, adenocarcinoma of the lung. Despite the administration of systemic chemotherapy, the disease underwent rapid progression and led to the patient’s death in a short period of time. Such an aggressive clinical course suggests that, in this specific case, KRAS dependency was the major genetic driver of poor prognosis. Direct deoxy ribonucleic acid (DNA) sequencing of the KRAS gene allows for the detection of novel KRAS mutations, and it might be advocated in patients with advanced non-small cell lung cancer in view of the emerging role of KRAS as a potential therapeutic target.
Keywords: DNA sequencing; enteric-type adenocarcinoma; gene mutation; KRAS; non-small cell lung cancer
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 28/07/2015; Received: 15/03/2015
Introduction
Mammalian cells express three closely related, small proteins of 189 amino acids with a molecular weight of 21 kDa (p21), termed KRAS, NRAS, and HRAS [1]. Localised in the inner plasma membrane, they function as GTPases that promote different types of ligand-mediated signal transduction pathways involved in cell proliferation, differentiation, and apoptosis [2]. RAS proteins cycle between two conformational states. One is when they are in the guanosine triphosphate (GTP)-bound active state, which is promoted by the guanine nucleotideexchange factors (GEFs); another one is when they bound to guanosine diphosphate (GDP), the inactive form, which is induced by the GTPase-activating proteins (GAPs).
Importantly, human cancers are preferentially associated with mutations in the KRAS gene, which almost invariably result into a constitutively active, GTP-bound RAS protein with pro-oncogenic effects [3]. In lung adenocarcinoma, somatic mutations of the KRAS gene are relatively frequent, occurring in approximately 11% and 26% of cases from Asian and Western patients, respectively [4]. In the majority of cases, these mutations are missense mutations that introduce an amino acid substitution at codon 12, 13, or, less frequently, 61 [3]. Here, we present the case of a patient with an enteric-type adenocarcinoma of the lung harboring a novel mutation at codon 22 of KRAS (Q22K) with concomitant KRAS polysomy.
Case report
A 74-year-old male with a 25 pack/year history of smoking was referred to the Medical Oncology of the Narni Hospital (TR, Italy) because of a voluminous right laterocervical lymphadenopathy. Two months earlier, he had started complaining of severe pain in the sacral region. An incisional biopsy of the enlarged lymph-nodal mass was positive for the presence of metastasis from poorly differentiated adenocarcinoma with signet ring cell features. A full-body computed tomography (CT) scan confirmed a 60 mm large laterocervical lymphadenopathy, also revealing a right lung lesion, a rib metastasis, and a voluminous neoplastic involvement of the first sacral vertebra (Figure 1), whose extension went beyond the spinal canal, infiltrating the radicular fibres. In addition, the patient underwent an endoscopic study of the gastroenteric tract with oesophagogastroduodenoscopy and colonoscopy, both tests excluding the presence of a primary gastrointestinal cancer. Histologic revision from a pathologist (EP) of the Perugia Hospital (PG, Italy) supported the diagnosis of pulmonary enteric-type adenocarcinoma (Figure 2).
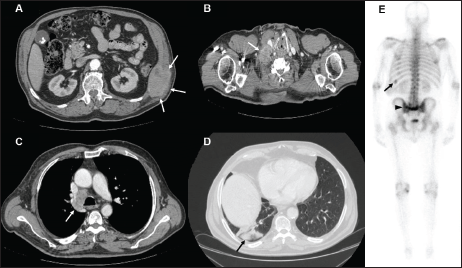
Figure 1. Contrast enhanced CT scan of case presentation. (A) osteolytic mass of the eleventh left rib (white arrows) invading surrounding soft tissues; (B) bulky lymph-nodal metastasis (white arrow) of the right posterior cervical space dislocating the right common carotid artery; (C) lower paratracheal, 4R according to IASLC, lymph-nodal metastasis (white arrow); (D) subpleural mass in the posterior segment of inferior right lobe; (E) Bone scintigraphy showing active pathological uptake of the left eleventh rib (black arrow) and of S2 vertebral body (black arrow head).
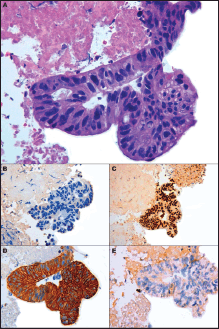
Figure 2. Microscopic presentation of the tumor showing enteric-type adenocarcinoma. (A) Cluster of neoplastic cells arranged in acinar pattern show atipical nuclei and clear cytoplasmatic droplets of mucin; (B) Immunohistochemically cells are negative to TTF-1; (C) Expression of CDX2 and (D) cytokeratin 7; (E) No immunoreactivity is evident for Cytokeratin 20. (A) Hematoxylin and Eosin, original magnification X40 - (B) X40 - (C) X20 – (D) X40 – (E) X40.
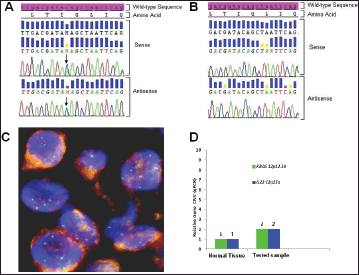
Figure 3. Biologic profile of the tumor. (A) Sequence analysis of the KRAS exon 2 performed on DNA extracted from microdissected area of paraffin-embedded tumor sections, shows a heterozygous point mutation: a C to A transversion (black arrow), resulting in a substitution of Glutamine, wild type (CAG) for Lysine (AAG) at codon 22 (p.Q22K, c.64C>A ). The mutation was named according to recommendations of the Nomenclature System for Human Gene Mutations. NCBI Reference Sequence: NM_004985.4 of KRAS gene was used as a reference; (B) Sequence analysis of the KRAS exon 2, performed on DNA extracted from peripheral white blood cells, shows the sequence wild type (CAG) resulting in normal glutamine at codon 22; (C) Dual colour FISH analysis (original magnification 60X) was performed using a CEP12 alpha satellite probe (12p11-q11) labelled in SpectrumOrange (Vysis, Downers Grove, IL. USA) and a BAC (Bacterial Artificial Chromosome) genomic probe RP11-707G18 (12p12.1) spanning an approximately 176 kb region encompassing the K-Ras gene, labelled in SpectrumGreen; (D) Representative data from KRAS and IL22 relative quantification (RQ) using TaqMan Copy Number Assays run on ViiA7 instrument. The number of copies of the KRAS and IL22 is determined by relative quantitation (RQ) using the comparative CT (ΔΔCT) method. This method measures the CT difference (ΔCT) between target and reference gene (RNase P), then compares the ΔCT values of test samples to a calibrator sample(s) known to have two copies of the target sequence. The copy number of the target is calculated to be two times the relative quantification.
Using somatic DNA extracted from the lymph-nodal biopsy, the sequence analysis for both epidermal growth factor receptor (EGFR) (exon 18 to 21) and KRAS (exon 2 and 3) genes was performed by nested polymerase chain reaction (PCR) and direct sequencing [5]. While no EGFR mutation was detected, a Q22K point mutation was described in the KRAS gene (Figure 3A). Such a mutation was not found in the peripheral white blood cells, thus indicating its somatic origin in the tumor (Figure 3B). Paraffin-embedded samples were then subjected to immunohistochemical (IHC) analysis for ALK (clone D5F3, cell signalling) and ROS1 (clone D4D6, cell signalling), which tested negative for both markers (not shown). To assess whether the KRAS Q22K mutation was accompanied by an increased KRAS gene copy number, KRAS FISH analysis was performed, which documented the presence of polysomy (≥ 4 copies in 10–40% of cells) for the KRAS gene (Figure 3C) [6]. In addition, quantitative polymerase chain reaction (qPCR) was carried out, which confirmed a copy number gain of 4 (Figure 3D) [7].
After receiving a single radiation dose of 8 Gy to the L4-S2 spinal tract, a decision was made to administer systemic chemotherapy with single-agent gemcitabine 1,000 mg/m2 days 1–8 every 21. A CT scan performed after three cycles of chemotherapy documented disease progression in the bone. On this basis, the patient was switched to second-line pemetrexed 500 mg/m2 every 21 days. However, after only two cycles of this regimen, the patient’s clinical conditions deteriorated rapidly, and he died because of progressive disease shortly thereafter.
Discussion
To our knowledge, this is the first report of a KRAS Q22K mutation in lung adenocarcinoma. This very rare mutation has been described only in a few other cancers, namely large/small intestine, haematopoietic and lymphoid, central nervous system (CNS), and pancreas, with a reported incidence that has been consistently lower than 1% for each of the aforementioned malignancies [8]. In the present case, a KRAS Q22K mutation was detected in an unusual variant of pulmonary adenocarcinoma, such as the enteric type. Importantly, the clinical presentation of the tumour was crucial in order to ascertain the primitive lung nature of the cancer (Figure 1).
KRAS Q22K mutation consists of a C to A transversion substituting lysine (AAG) for normal glutamine (CAG) at the 22nd amino acid residue of RASp21, which is structurally located between the GTPase domain and the effector domain binding GAPs [9]. As a result, the affinity of RASp21 for guanine nucleotide is influenced, which allows KRAS Q22K mutant to be stabilised into a constitutively activated GTP-bound form. Accordingly, preclinical studies have shown that cell lines expressing the KRAS Q22K mutation possess high in vivo oncogenic potential, higher than that of wild-type KRAS [9]. This finding is in line with the evidence that non-small cell lung cancer (NSCLC) cell lines transfected with KRAS Q22K mutants show elevated levels of RAS-GTP and downstream phosphorylated ERK, compared with wild-type KRAS [10].
Inconclusive data have been reported in the literature about the role of KRAS mutation as ‘driver’ genetic alteration in advanced NSCLC [11, 12]. Current knowledge only suggests that the presence of KRAS mutation is a negative predictor for treatment with an EGFR-TKI [13]. However, the heterogeneity of KRAS mutations in NSCLC limits the further applicability of this finding [14]. Nevertheless, in this specific case, the ‘driver’ role of KRAS Q22K mutation can be deduced from a few reasons. First, the mutation was confirmed to be somatic (Figure 1B). Secondly, some other ‘driver’ genetic alterations were excluded (i.e., EGFR, ALK, and ROS1), thus suggesting the presence of KRAS mutation as a single-mutated oncogene. Also, KRAS FISH and qPCR analyses showed polysomy of the KRAS gene, and preclinical models of KRAS Q22Kmutant tumors have shown even enhanced tumorigenicity in the presence of increased KRAS gene copy number (either KRAS amplification or KRAS polysomy) [9]. Accordingly, a worse prognosis has been suggested for patients who harbour a KRAS mutation simultaneously with an increased KRAS gene copy number > 3 [15]. Finally, as shown by the sequencing electropherogram (Figure 1A), KRAS mutant allele peak was equal to that of wild-type KRAS, which is in line with a previous report suggesting worse prognosis in the presence of KRAS-mutant allele greater than or equal to wild-type allele [16]. Taken together, these data strongly suggest that, in this specific case, the proliferation and survival of the tumour was largely sustained by KRAS Q22K mutation and simultaneous KRAS polisomy.
Conclusions
The present case unveils a novel KRAS Q22K mutation in lung adenocarcinoma and suggests the potential driver role of this specific KRAS-mutant genotype when associated with increased KRAS gene copy number. Finally, it supports direct DNA sequencing as the ‘gold standard’ for the detection of KRAS mutations in NSCLC, since it allows the uncovering of uncommon mutations as opposed to ‘target’ methods (e.g., Pyrosequencing, TheraScreen), which reveal only predetermined KRAS gene mutations. This is of particular relevance in view of the ongoing clinical trials that are testing the effectiveness of molecules aimed at inhibiting the RAS/RAF/MEK/ERK pathways [17, 18], which could be offered as an alternative treatment approach to patients biologically selected for KRAS mutation.
Conflicts of interest
The authors have no conflict of interest to declare.
Acknowledgments
The authors thank the patient for providing written informed consent for the publication of this case.
Supported by the Italian Association for Cancer Research (AIRC).
References
1. Platz A et al (2008) Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site Mol Oncol 1 395–405 DOI: 10.1016/j.molonc.2007.12.003
2. Stephen AG et al (2012) Dragging ras back in the ring Cancer Cell 25 272–281 DOI: 10.1016/j.ccr.2014.02.017
3. Prior IA, Lewis PD and Mattos C (2012) A comprehensive survey of Ras mutations in cancer Cancer Res 72 2457–2467 DOI: 10.1158/0008-5472.CAN-11-2612 PMID: 22589270 PMCID: 3354961
4. Dearden S et al (2013) Mutation incidence and coincidence in nonsmall-cell lung cancer: meta-analyses by ethnicity and histology (mutMap) Ann Oncol 24 2371–2376 DOI: 10.1093/annonc/mdt205 PMID: 23723294 PMCID: 3755331
5. Ludovini V et al (2012) Optimization of patient selection for EGFR-TKIs in advanced non-small cell lung cancer by combined analysis of KRAS, PIK3CA, MET, and non-sensitizing EGFR mutations Cancer Chemother Pharmacol 69 1289–1299 DOI: 10.1007/s00280-012-1829-7 PMID: 22302407
6. Smith G et al (2010) Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours—implications for personalised cancer medicine Br J Cancer 102 693–703 DOI: 10.1038/sj.bjc.6605534 PMID: 20147967 PMCID: 2837563
7. Bardelli A et al (2013) Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer Cancer Discov 3 658–673 DOI: 10.1158/2159-8290.CD-12-0558 PMID: 23729478 PMCID: 4078408
8. COSMIC—Catalogue of somatic mutations in cancer Available at: http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/ [accessed 1.1.15]
9. Tsukuda K et al (2000) A novel activating mutation of the K-ras gene in human primary colon adenocarcinoma Biochem Biophys Res Commun 278 653–658 DOI: 10.1006/bbrc.2000.3839 PMID: 11095964
10. Janakiraman M et al (2010) Genomic and biological characterization of exon 4 KRAS mutations in human cancer Cancer Res 70 5901–5911 DOI: 10.1158/0008-5472.CAN-10-0192 PMID: 20570890 PMCID: 2943514
11. Meng D et al (2013) Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: a systematic review with meta-analysis Lung Cancer 81 1–10 DOI: 10.1016/j.lungcan.2013.03.019 PMID: 23608713
12. Metro G et al (2014) Clinical outcome with platinum-based chemotherapy in patients with advanced nonsquamous EGFR wild-type non-small-cell lung cancer segregated according to KRAS mutation status Clin Lung Cancer 15 86–92 DOI: 10.1016/j.cllc.2013.08.002
13. Mao C et al (2010) KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies Lung Cancer 69 272–278 DOI: 10.1016/j.lungcan.2009.11.020
14. Metro G et al (2012) Impact of specific mutant KRAS on clinical outcome of EGFR-TKI-treated advanced non-small cell lung cancer patients with an EGFR wild type genotype Lung Cancer 78 81–86 DOI: 10.1016/j.lungcan.2012.06.005 PMID: 22770374
15. Sasaki H et al (2011) Evaluation of Kras gene mutation and copy number gain in non-small cell lung cancer J Thorac Oncol 6 15–20 DOI: 10.1097/JTO.0b013e31820594f0
16. Chiosea SI et al (2012) KRAS mutant allele-specific imbalance in lung adenocarcinoma Mod Pathol 24 1571–1577 DOI: 10.1038/modpathol.2011.109
17. Jänne PA et al (2013) Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study Lancet Oncol 14 38–47 DOI: 10.1016/S1470-2045(12)70489-8
18. Blumenschein GR et al (2013) MEK114653: a randomized, multicenter, phase II study to assess efficacy and safety of trametinib (T) compared with docetaxel (D) in KRAS-mutant advanced non–small cell lung cancer (NSCLC) J Clin Oncol 31(suppl) abstract 8029

