Primed for cancer: Li Fraumeni Syndrome and the pre-cancerous niche
Pan Pantziarka
The George Pantziarka TP53 Trust, London KT1 2JP, UK
Anticancer Fund, Brussels, 1853 Strombeek-Bever, Belgium
Correspondence to: Pan Pantziarka. Email: anticancer.org.uk@gmail.com
Abstract
The complex relationship between tumour and stroma is still being elucidated but it is clear that cancer is a disease of more than just malignant cells. However, the dominant focus of our current understanding of Li Fraumeni Syndrome (LFS) remains on the function of p53 as ‘guardian of the genome’. Recent evidence shows that the TP53 gene is at the nexus of a wider range of functions, including aspects of cellular metabolism, aging and immunity. Incorporating this broader picture of the role of TP53 together with our understanding of the role of the host microenvironment in cancer initiation and progression gives a more nuanced picture of LFS. Furthermore, there is clinical evidence to suggest that the host environment in healthy individuals with LFS already includes some of the features of a ‘pre-cancerous niche’ that makes cancer initiation more likely. It is suggested, finally, that there are pharmacological interventions capable of altering this pre-cancerous niche, thus potentially reducing the cancer risk in individuals with LFS.
Keywords: TP53, Li Fraumeni Syndrome, cancer predisposition, pre-cancerous niche
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 21/05/2015; Received: 06/02/2015
Introduction
Li Fraumeni Syndrome (LFS) is a rare autosomally dominant genetic syndrome associated with a highly elevated risk of developing cancer. First identified in a group of families with an unusually increased pattern of cancer incidence by Frederick Li and Joseph Fraumeni in 1969, the syndrome is characterised by early development of breast cancer and a range of sarcomas, adrenocortical carcinoma, gliomas, leukaemias, and other cancers, particularly in children and early adulthood [1, 2]. LFS is associated with germ-line TP53 tumour suppressor gene mutations, although it is estimated that around 20–30% of individuals diagnosed with the condition do not carry such a mutation [3, 4].
Breast cancer, very often pre-menopausal, is the most common tumour in women with LFS, with an estimated risk of 49% by the age of 60, with one recent study finding the median age of diagnosis as 32 years (range 22–46) [5]. Around 50% of sufferers will develop cancer before the age of 30 [6], while the life-time risk of developing cancer has been estimated at 70% for males and 100% for women [7]. The most common cancers are breast cancer (27%) and soft-tissue and bone sarcomas (25%) [6].
A number of different diagnostic criteria exist for LFS, and in addition to ‘classical LFS’, there is a recognised Li Fraumeni-like (LFL) Syndrome, which has less stringent diagnostic and testing criteria. The different criteria are discussed in [6] and [8]. The management of LFS post-diagnosis is broadly consistent globally. There is an emphasis on active surveillance, with new protocols being trialled in different parts of the world (for example NCT01464086 and NCT01737255), and women are offered the opportunity to have risk-reducing double mastectomy. Many of the new surveillance protocols under investigation focus on the use of regular whole-body MRI, ultrasound and other clinical investigations that do not impose additional exposure to ionising radiation. Results from some studies of such protocols have shown that active surveillance can lead to significant survival benefits, for example one small non-randomised cohort study found that patients in the active surveillance group had a 100% three-year survival, in contrast to a 21% survival in a matched non-surveillance group [9]. Aside from the option of mastectomy for women, there are no other active cancer-prophylactic measures in clinical use or currently being trialled.
Whilst TP53 is the gene most associated with LFS, there is evidence that some individuals harbour mutations in related genes. For example analysis of non-TP53 mutant LFS families showed a greater incidence of a single-nucleotide polymorphism 309 in the MDM2 gene than in the general population [10]. MDM2 is a key negative regulator of TP53, and this particular polymorphism results in increased expression of MDM2 and a consequent attenuation of the p53 pathway. While there is animal data suggesting that MDM2 polymorphisms may be associated with elevated cancer incidence [11], the prognostic significance of mutations in other genes in the TP53 pathway have yet to be established in humans.
LFS is not the only disease associated with TP53 abnormalities. There is increasing evidence of a role of defective p53 activity in the pathophysiology of a number of other rare genetic conditions, including Diamond-Blackfan anemia (DBA), CHARGE syndrome and ATR-Seckel syndrome. In these diverse syndromes, which are associated with a range of congenital abnormalities, altered p53 activity is implicated in certain stem cell niches [12–14].
Because of the centrality of the p53-associated tumour suppressor pathway, carcinogenesis in LFS is viewed primarily through the lens of genetic damage. It is assumed that in LFS cancer develops because of additional genetic damage to the TP53 gene causing loss of heterozygosity (LOH) and/or gain of function (GOF), leading to malignant transformation [15–17]. In effect cancer incidence in LFS is due to the ‘two-hit’ model first proposed by Knudson based on analysis of hereditary transmission of retinoblastoma [18]. One immediate consequence of this model is a caution in treating LFS patients with radiotherapy or limiting exposure to ionising radiation during diagnostic investigation [6, 19].
In recent years our understanding of the role of p53 has expanded considerably, and now includes key functions in aging [20, 21], cellular metabolism [22, 23], regulation of homeostasis [24] and immune function [25, 26]. Research has also focused on the non-cell-autonomous functions of p53 which can suppress tumourigenesis by the promotion of an anti-tumour microenvironment [27, 28]. By the same token, just as the apoptotic program can be subverted, so too can the non-cell-autonomous functions be subverted to promote a pro-tumourigenic microenvironment. This complex interplay between p53 and the microenvironment is central to the thesis outlined in this paper.
We are also beginning to explore the complex landscape of TP53 mutations and the role this plays in cancer initiation, progression and treatment response [29–31]. Similarly the study of the impact of p53 isoforms is at an early stage of development, but it is already clear that different isoforms can radically alter transcriptional activity and core functionality, again differentially impacting cancer initiation, progression and response to treatment [32, 33].
In the same period we have also seen an increased understanding of the role of the microenvironment in cancer initiation, progression and metastasis [34–36]. Where once cancer was seen as primarily a disease of mutated cells, it is now increasingly being viewed as a dynamic evolutionary system that incorporates stromal cells, immune infiltrates, tumour vasculature and heterogenic clonal subpopulations of cancer cells [37, 38].
These new insights into the functionality of p53 and the broader understanding of tumour biology have yet to be integrated into our understanding of cancer initiation in individuals with LFS. This paper will attempt to relate this new knowledge with a number of clinical features common in individuals with LFS and to shed light on the implications these might have on cancer risk. In particular it will propose a more nuanced view of carcinogenesis in LFS, suggesting that there are important additional factors over and above genetic ablation of p53 tumour suppressor function.
The importance of the host environment
Stephen Paget, building on work by Ernst Fuchs, proposed the ‘seed and soil’ hypothesis in 1889 to explain the tendency of breast cancers to metastasise preferentially to certain tissues [39]. He suggested that the ‘soil’ (host environment) was as important as the ‘seed’ (the migrating tumour cell) in the complex process of metastasis, and that certain tissues (bone, brain, liver) provide a more hospitable environment for cancer cells to take ‘root’ and produce viable tumours. While the hypothesis directs attention to the network of relationships between migrating tumour cells and the various elements of the host environment, it is also a reminder that there is much still to be explained about the process of metastatic spread of disease. Many key controversies remain to be resolved, for example do cancer cells acquire metastatic potential early or late in the process of tumourigenesis? Where initial theory suggested that metastatic potential was acquired at a late stage in primary cancer growth, driven by the accumulation of genetic changes and clonal evolution, newer theories suggest that it is in fact a property of certain subtypes of cancer cells and that these cells have an intrinsic ability to disseminate to and proliferate in distant sites [40]. Of more consequence to the thesis outlined in this paper, there has been a greater interest in understanding the nature of the ‘soil’, in particular in understanding what it is that makes certain tissues more conducive to tumour growth than others.
An important stream of work has explored the idea of a ‘pre-metastatic niche’, an idea first elucidated by David Lyden and colleagues in 2005–2006 [41, 42]. In this model it is not the intrinsic properties of the migrating tumour cell that determines the success or otherwise of the metastatic process, but the properties of the host microenvironment in which the tumour cell lodges. A permissive environment will provide the growth factors, nutrient flow and support systems that enable the tumour cell to proliferate and grow into a viable metastatic nodule. In contrast, an inhospitable host environment will not provide the factors that the tumour cell needs to survive and proliferate, and therefore no metastatic growth will form regardless of the properties of the tumour cell. Furthermore, it appears that there are certain tissues and microenvironments which are more likely to harbour these pre-metastatic niches, and that these tissues correspond to the most likely sites of metastatic spread for different cancer types.
Analysis of the process of metastasis shows that certain cell types, in particular hematopoietic progenitor cells (HPC) expressing VEGFR-1 become clustered at sites prior to the arrival of tumour cells, a finding confirmed in breast cancer patients in addition to murine models [41]. The clustering of these VEGFR-1 HPCs initiates a cascade that includes the expression of integrins (including VLA-4), matrix metalloproteinase 9 (MMP9), and downstream expression of VEGF-A and fibronectin expression in resident fibroblasts. Furthermore, in addition to local tissue remodelling, there is evidence that these clusters promote the chemoattraction and attachment of circulating tumour cells and endothelial progenitor cells (EPC), which are essential for angiogenesis to take place [43].
Formation of these clusters of VEGFR-1 HPCs is triggered by the release of factors from the primary tumour with homing to specific sites related to ligand-receptor pairing. For example breast cancer cells expressing the chemokine receptor CXCR4 home to organs with high levels of its ligand SDF-1 (stromal derived factor-1), for example the bone marrow, lungs and liver [43]. In a murine model it has been shown that the inflammatory chemoattractants S100A8 and S100A9 released from primary B16 melanoma tumours pre-condition the lungs, create pre-metastatic niches and attract both tumour and inflammatory cells [44].
McAllister and colleagues investigated mechanisms by which primary tumours in a mouse model are able to ‘condition’ distant tissues to create conditions in which otherwise indolent tumour cells became activated through the recruitment of a reactive stroma, a process they termed systemic instigation [45, 46]. However, both the primary tumour and the indolent tumour cells were implanted in the animal model used, and therefore this conditioning of a metastatic niche was co-incident with tumour cell inoculation. Similarly Massagué et al identified cancer-cell expression of tenascin-C (TNC), an extracellular matrix (ECM) protein of stem cell niches, as essential for the establishment of breast cancer metastases in the lungs [47]. Initial expression of TNC by breast cancer cells was required to maintain viability in the lung parenchyma, through up-regulation of Wnt and Notch signalling, until stromal expression of TNC by infiltrating myofibroblasts or other stromal sources accumulated in the expanding nodules to provide a supportive metastatic stem cell niche.
Liu and colleagues used a mouse of breast cancer metastasis to the lungs to show that primary tumours were able to induce inflammatory changes via release of VEGF and consequent recruitment of bone-marrow-derived cells in the lungs [48]. Furthermore VEGF induced prostaglandin E2 (PGE2) production in pulmonary endothelial cells and enhanced the adhesion of injected circulating tumour cells. Analysis showed that the sites of inflammatory response in the lungs preferentially harboured injected tumour cells, showing that the primary tumour was able to induce pre-metastatic niche formation.
A recent review by McAllister and Weinberg summarises additional evidence showing that primary tumours can condition distant tissues, including discussion of pre-metastatic niche creation [49].
A key feature of this pre-metastatic niche is the inclusion of multiple stromal cell types, including fibroblasts, macrophages and other immune cells, HPCs and EPCs. These are, of course, also many of the components of the primary tumour microenvironment (TME) [50]. Increasingly our view of cancer is that of an evolving and dynamic ecosystem that incorporates tumour and non-tumour cells in complex patterns of competition and cooperation. No longer focused purely on the genomic changes within the tumour cell, viewing cancer as an ecosystem redirects attention to those factors in the microenvironment which support survival, proliferation and ultimately the metastatic process [37, 51, 52].
Significantly, an understanding of the importance of the TME to disease progression, resistance to treatment and metastatic dissemination also creates new opportunities for therapeutic intervention [53]. Altering aspects of the TME, by targeting specific populations of stromal cells or signalling pathways, can interfere with the ecological balance within the tumour and render cancer more immunogenic, less resistant to chemotherapy or radiation and so on.
The pre-cancerous niche
An extension of the idea of the pre-metastatic niche to primary disease has also been discussed [54]. A multi-step model of primary carcinogenesis has been proposed, starting with niche construction leading to expansion and maturation. According to this model, tumourigenesis cannot take place without the existence of the pre-cancerous niche, regardless of the accumulated genetic mutations in a transformed cell. Numerous experiments have indeed shown that malignant cells transplanted into non-transformed tissues (in other words in tissues lacking the pre-cancerous niche) experience loss of malignant behaviour [55–57]. Initiation of niche construction may have multiple physiological causes, including the local action of carcinogenic agents, tissue injury, infection or aging [54].
A common feature of the pre-cancerous niche, independent of the initial cause, is the presence of chronic inflammation. The link between cancer and inflammation is well-characterised, and it is being increasingly recognised as causative rather than a by-product of disease progression [58–60]. Chronic inflammation is associated with cancer incidence related to bacterial and viral infections, chronic disease (liver cirrhosis, diabetes), tobacco-smoke inhalation, obesity and aging. It is also associated with the expansion and maturation of the pre-cancerous niche, with the recruitment of inflammatory cells, activation and remodelling of stromal components (particularly fibroblasts), release of pro-angiogenic factors and the homing of transformed cells [35, 36]. Tumour formation occurs through the co-evolution of the niche and homed transformed cells, creating a tumour microenvironment that supports further proliferation and invasion of the complex and co-evolving mass.
It is clear that this complex process, which has yet to be fully elucidated, incorporates many of the hallmarks of cancer [61]. It is also clear that it is a challenge for researchers in LFS to incorporate this complex picture into our understanding of cancer initiation in people with this condition.
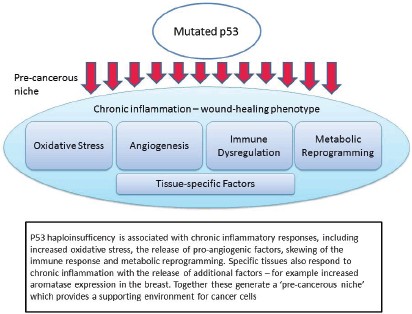
Figure 1. Mutated p53 function leads to the creation of a pre-cancerous niche.
TP53 and the pre-cancerous niche
It is our contention that people with LFS are ‘primed’ for cancer initiation because a number of the key drivers of niche initiation are a feature of the non-cancerous LFS host. That is, they are at greater risk of cancer initiation because of more than defective tumour suppressor activity. In other words many of the other functions of the p53 network are also important drivers of cancer risk. Equally important, it is possible that these other functions, many of which directly impact the formation of the pre-cancerous niche, are amenable to drug targeting in a way that the pro-apoptotic function of p53 signalling is not.
The non-apoptotic functions which are important in this context are: chronic inflammation and oxidative stress; pro-angiogenic signalling; immune dysregulation; metabolic reprogramming; and tissue-specific interactions (Figure 1). Each of these will be discussed in turn, noting both the evidence from recent studies in p53, but also relevant clinical evidence from LFS patients or appropriate animal models.
Chronic inflammation and oxidative stress
The role of chronic inflammation in cancer initiation and progression is well-known and well-characterised [46]. Inflammatory cells, including neutrophils, monocytes, macrophages, mast cells and lymphocytes, are recruited to the site of the inflammatory response. Histamine is released causing increased vasodilation. Pro-angiogenic factors are released and extensive remodelling of the tissues takes place. Reactive oxygen species (ROS), produced by multiple inflammatory cell types, further drive inflammation in a positive feedback loop [59]. The p53 network is activated during this inflammatory state, most likely in direct response to the cellular stresses invoked by elevated ROS levels [63, 64].
However, there is also evidence that loss of p53 function can itself act as a driver of inflammation in an NF-kB-dependent manner [65]. Significantly, this study showed that loss of p53 alone was insufficient to cause tumourigenesis. The relationship between increased inflammation, oxidative stress and p53 is apparent in numerous pre-cancerous or inflammatory conditions [66–69]. Furthermore, there is also evidence that cancer-free LFS sufferers exhibit clinical signs of increased levels of oxidative stress compared to a paired group of family members without TP53 mutations [70].
Angiogenesis
Tumour neo-angiogenesis is an important rate-limiting step in tumour growth and progression. In terms of the pre-cancerous niche and LFS, there are two elements that are of interest in the development of a pro-angiogenic environment. In the first case the increased oxidative stress associated with chronic inflammation acts to induce angiogenesis as part of a ‘wound healing’ response [71, 72]. However, p53 is also known to have an influence on angiogenesis via thrombospondin-1 (TSP-1), which acts as an endogenous anti-angiogenic factor [73, 74]. Loss of p53 leads to down-regulation of TSP-1 which correlates to increased expression of VEGF and other pro-angiogenic signals [75, 76].
There is also some evidence that a number of micro-RNAs, some of which are involved in the tumour suppressive and stress response functions of p53, have an influence on angiogenesis [77]. For example miR-107 mediates the p53 response to hypoxia by suppressing the expression of hypoxia inducible factor-1β (HIF-1β), which in turn down-regulates expression of VEGF [78]. Impaired transcriptional activity in mutant p53 may therefore lead to loss of anti-angiogenic activity and increased expression of VEGF and angiogenesis [79].
The evidence from fibroblasts derived from LFS patients confirms that loss of the wild-type p53 allele is sufficient to decrease TSP-1 expression and an increase in VEGF [73]. A step-wise process may take place whereby additional changes, such as oncogene activation, may lead to a more fully pro-angiogenic phenotype [80]. Additionally it should be noted that there is evidence that some gain-of-function (GOF) TP53 mutations (including R175H and R273H common in people with LFS) have been shown to have tumour angiogenesis promoting activity [81, 82].
Immune Dysregulation
Little work has been done to investigate the effect that lack of wild-type p53 in stromal cells has on immune response to tumour growth. One notable exception compared the rate of tumour development in mice with different p53 status, observing that implanted B16F0 melanoma tumours grew at a faster rate in mice lacking wild-type p53 [83]. Furthermore, the effect was not apparent in severe combined immunodeficiency mice, suggesting that this difference in tumour growth is immune-related. Tumours were also implanted in wild-type mice along with mesenchymal stem cells (MSC), a major part of the microenvironment, with differing p53 status. Wild-type mice co-implanted with stromal cells lacking p53 developed larger tumours than mice with wild-type cells in the stroma. The p53-deficient MSC cells inhibited T-cell function, over-expressed inducible Nitric Oxide synthase and generated an immunosuppressive microenvironment conducive to tumour growth.
There is also an influence on immune response via p53 regulation of toll-like receptor (TLR) expression [84]. TLRs, the pattern recognition receptors involved in the innate immune response, are able to respond to both exogenous and endogenous ligands, are also now recognised to have a role in adaptive immunity [85]. Furthermore, TLRs, which are now known to be expressed by cancer cells in addition to a range of non-cancer cells [86], have a complex role in cancer, with both pro- and anti-cancer activity [87, 88]. There is some evidence that TLR signalling plays an active role in carcinogenesis during chronic inflammation, particularly with respect to facilitating escape from immune surveillance, release of pro-inflammatory cytokines and chemokines and tumour angiogenesis [86, 89]. TLR expression is modulated by p53 activation, and there is evidence that TP53 mutants, including a number associated with germ-line mutations, can differentially impact TLR expression in response to cellular stressors [84, 90].
There is also emerging evidence that p53 has a role in the regulation of PDL1 (programmed death ligand 1), a transmembrane protein expressed by tumour cells that is the target of intense scientific interest in relation to its role in disarming anti-tumour immune responses in a range of malignancies [91]. Data presented by Cortez and colleagues at AACR 2015 showed that PDL1 is regulated by p53 via miR-34a in NSCLC [92].
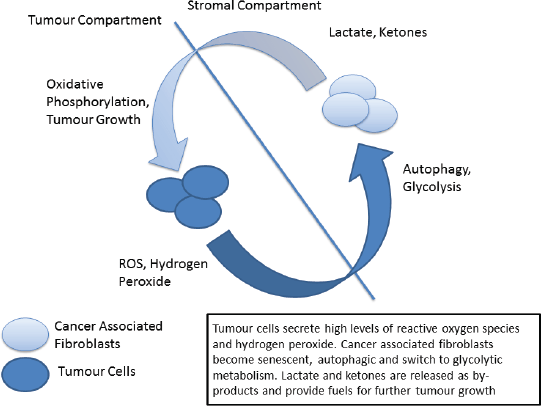
Figure 2. The ‘reverse Warburg’ phenotype.
Metabolic Reprogramming
Metabolic reprogramming is another of the hallmarks of cancer in which p53 plays a central role [93, 94]. The traditional view, termed the Warburg Effect, focuses on the increased metabolic needs of malignant tissues and a consequent metabolic switch to glycolysis in tumours. The Warburg effect manifests itself as an increase in glucose metabolism by tumour cells, the generation of lactate as a by-product and an increase in acidity and hypoxia, both driving clonal evolution towards a more malignant phenotype. However, in recent years this view has been challenged by the emergence of the ‘reverse Warburg effect’ hypothesis in which distinct metabolic compartments exist within the tumour and stromal cell populations [95, 96]. In this model increased oxidative stress drives cancer associated fibroblasts to switch to glycolytic metabolism, producing lactate and other by-products which are metabolised by tumour cells – effectively creating a metabolic shuttle between tumour and stroma (Figure 2) [97]. Analysis of patient samples in a variety of cancer types has indicated the presence of both ‘Warburg’ and ‘reverse Warburg’ phenotypes, suggesting that cancer cells are metabolically plastic and can adapt and change during disease progression [98, 99]. Of note the ‘reverse Warburg’ phenotype has also been detected in osteosarcoma, one of the ‘signature’ cancers associated with LFS [100].
It has previously been proposed that cancer initiation in LFS is related to this ‘two compartment model’ of tumour metabolism [101]. Briefly, p53 signalling in response to increased oxidative stress can trigger cellular autophagy in fibroblastic cells, shifting their metabolism towards glycolysis. The increase in secretion of lactate drives further changes in the microenvironment, producing an immunosuppressive and pro-tumour environment. Cells may also react to environmental and metabolic stresses by undergoing senescence, again mediated by p53, eventually becoming immortalised and/or undergoing malignant transformation. When these cells become transformed it is in a hospitable microenvironment that has been ‘primed’ for cancer initiation.
A key marker for the ‘reverse Warburg’ or ‘two-compartment’ phenotype is loss of stromal cav-1 expression [102], and this finding has been confirmed in people with LFS compared to non-affected family members [103].
Tissue-specific factors
While the factors listed above are common to pre-cancerous niches in multiple tissue types, there are also factors which are specific to some tissues and not others. This accords with the observations both in primary disease and in the pattern of metastatic spread preferentially to given tissues, for example bones, lungs and liver in breast cancer. In particular cancer incidence in LFS is more strongly associated with certain tissues (e.g. breast, sarcomas, adrenocortical carcinoma) than with others (e.g. lung, bladder). It is hypothesised that this pattern of primary tumour incidence is related to secreted factors in specific tissues which contribute to pre-cancerous niche formation.
An analysis of the breast cancer phenotype in women with LFS found that 84% of invasive tumours were hormone responsive (ER and/or PR), with a majority of these also being positive for Her2/neu, figures which are higher than for a comparable non-LFS population [5]. Evidence that this may be related to factors in stromal tissues comes from a recent analysis of the relationship between stromal aromatase expression, which is associated with ER breast cancer, and p53 in breast tissue [104]. The authors analysed breast adipose tissue to examine the relationship between aromatase expression, prostaglandin E2 (PGE2) and p53. Results indicated a feedback loop between p53 and the expression of aromatase and that low p53 expression caused increased aromatase. This relationship between aromatase and p53 was shown to be mediated by PGE2, which down-regulates p53 expression. Significantly, analysis of tissue samples from women with LFS showed statistically significantly higher levels of aromatase in tumour-associated stromal adipose tissue compared to non-LFS women.
A different view of cancer initiation
The pre-cancerous niche is, as we have seen, a hospitable and conducive environment in which tumours can take root and prosper. A germ-line TP53 mutation leads to many of the key features of such a niche being expressed in otherwise healthy and non-cancerous LFS individuals. Many of the features of this niche environment are self-reinforcing or coupled in positive feedback loops such that inflammatory and pro-tumour signalling is perpetuated leading to chronic inflammation.
The question that arises is whether this pre-cancerous niche is itself a driver of carcinogenesis in people with LFS. Again, the lack of functioning wild-type p53 may conspire to initiate transformation and carcinogenesis. It is known that telomere length is shorter in people with LFS compared to non-affected family members [105, 106], and that this may be related to the age of cancer onset [107, 108]. Progressive shortening of telomeres, a process that is exacerbated by oxidative and other cellular stresses, eventually leads to ‘telemore crisis’ and consequent DNA damage. At this stage the p53 damage response may lead to senescence or apoptosis, and indeed there is evidence that cells from LFS patients display greater levels of DNA damage (chromosomal instability, senescence etc) [109–111].
We may speculate, therefore, that the combination of lack of wild-type p53 and increased oxidative stress may therefore cause the subsequent genetic damage leading to cellular transformation and cancer initiation. One possible scenario is that the increased oxidative stress in the pre-cancerous niche causes telomere shortening in adjacent non-stromal cells, leading to crisis and initiating a sequence that eventually leads to malignant transformation (Figure 3). Transformed cells in contact with the pre-cancerous niche find a hospitable ‘soil’ in which they ‘seed’ tumour growth.
This differs from the conventional view of carcinogenesis in LFS being caused by a random ‘second hit’ mutation leading to cancer. Instead it suggests very strongly that the phenotypic features of the LFS host which are caused by the germ-line mutation in TP53 creates a set of pre-cancerous niches which may act to cause the ‘second hit’ or the switching on of oncogenic pathways.
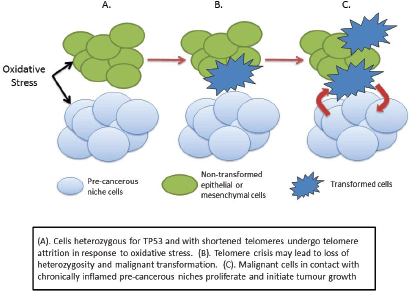
Figure 3. Cancer initiation in LFS as a consequence of haploinsufficiency of p53.
Supporting evidence for this hypothesis comes from a series of animal experiments in a mouse model of LFS [112]. Heterozygous and wild-type Trp53 mice were treated with either surgical implantation of a foreign object to induce chronic inflammation, or a sham operation. Thirty of 38 (79%) heterozygous mice developed sarcomas around the implant at a mean age of 46 weeks, compared to one (10%) of the wild-type mice at 56 weeks. No sarcomas developed at the sites of sham operation. Two of 10 (20%) control heterozygous mice (no implant) also developed sarcomas, but at a mean age of 80 weeks. In 90% of implant-induced sarcomas, loss of heterozygosity was observed suggesting a causative effect from the chronic inflammation induced by the implant. This is in contrast to the work of Lyden and colleagues in that this was a model of primary carcinogenesis rather than metastatic spread through the creation of a pre-metastatic niche by the primary tumour.
Of note it is the phenotypic features of the host which are important drivers of this process. These features may also be present in people diagnosed with LFS or LFS-like conditions in which there is no TP53 mutation detected. It is possible that a similar process takes place in other cancer predisposition syndromes in addition to LFS. There may be a number of different genetic drivers which can lead to the creation of pre-cancerous niches in Cowden Syndrome (associated with the PTEN tumour suppressor), Peutz–Jeghers Syndrome (associated with STK11/LKB1 gene) and other such conditions. Further investigation of this hypothesis is warranted as it may mean that similar clinical strategies may apply to a range of syndromes normally viewed and treated in isolation.
Conclusion
In widening our view of cancer initiation in LFS to include the non-apoptotic functions of the TP53 gene we can see that the microenvironment assumes a much greater significance. This has important clinical implications for people with LFS for it is known that some of the features of the micro-environment may be more easily amenable to drug targeting than p53 itself. Indeed some pre-clinical work in animal models has already produced intriguing results.
For example, focusing purely on metabolic plasticity, Komarova and colleagues showed that the mTOR inhibitor rapamycin, which can act to inhibit senescence, increased the life-span and delayed tumourigenesis in mice bearing heterozygous p53 mutations [113]. In terms of the ‘two compartment’ model of tumour metabolism, inhibiting senescence in stromal cells may lead to fewer cells switching to glycolysis and thus depriving coupled tumour cells of lactate and other high-energy fuels. Similarly, caloric restriction in a mouse model of LFS also delayed tumour onset in adult mice [114], which may be related to reduced supply of glucose feeding glycolytic cells.
However, there are other features of the pre-cancerous niche which can also be targeted. Reductions in chronic inflammation or oxidative stress may also be possible. For example the anti-diabetic drug metformin has pleiomorphic effects which may be suitably exploited to attack the pre-cancerous niche in multiple ways:
• Reduction in oxidative stress
• Reduction in chronic inflammation
• Action on metabolic pathways in tumour and/or stromal cells
• Reduction of hepatic glucose production
• Pro-apoptotic effects in some cancers
• Increase in anti-tumour immunity [115]
A range of other drugs, many of which are being actively investigated as possible repurposed anti-cancer drugs [116] also show some promise, including aspirin and other NSAIDs and the lipophilic statins. Other strategies may include dietary and other interventions to reduce oxidative stress, chronic inflammation or increased telomere attrition.
Investigation of the cancer-preventative effect of these drugs in relevant animal models is warranted, as is further investigation of the features of the pre-cancerous niche in people with LFS. It may be that there is prognostic significance in measures of oxidative stress, level of cav-1 expression or other biomarkers related to chronic inflammation.
Clinical trials of cancer prevention in LFS are problematic given the relatively large sample sizes required to show efficacy of any interventions. However, if the pre-cancerous niche hypothesis proves to be equally applicable to other cancer predisposition syndromes then an increased pool of patients may mean that clinical trials may become more feasible. It is suggested therefore, that further investigation of this hypothesis be undertaken in other cancer predisposition syndromes.
Competing interests
The author declares that there are no competing interests. This research was funded by the George Pantziarka TP53 Trust.
References
1. Li FP and Fraumeni JF (1969) Soft-tissue sarcomas, breast cancer, and other neoplasms A familial syndrome? Ann Intern Med 71(4) 747–752 DOI: 10.7326/0003-4819-71-4-747 PMID: 5360287
2. Malkin D (2011) Li-Fraumeni syndrome Genes Cancer 2(4) 475–484 DOI: 10.1177/1947601911413466 PMID: 21779515 PMCID: 3135649
3. Malkin D, Li FP and Strong LC et al (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms Science 250(4985) 1233–1238 DOI: 10.1126/science.1978757 PMID: 1978757
4. Nichols KE, Malkin D and Garber JE et al (2001) Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers Cancer Epidemiol Biomarkers Prev 10(2) 83–87 PMID: 11219776
5. Masciari S, Dillon DA and Rath M et al (2012) Breast cancer phenotype in women with TP53 germline mutations: a Li-Fraumeni syndrome consortium effort Breast Cancer Res Treat 133(3) 1125–1130 DOI: 10.1007/s10549-012-1993-9 PMID: 22392042 PMCID: 3709568
6. McBride KA, Ballinger ML and Killick E et al (2014) Li-Fraumeni syndrome: cancer risk assessment and clinical management Nat Rev Clin Oncol DOI: 10.1038/nrclinonc.2014.41 PMID: 24642672
7. Chompret A, Brugières L and Ronsin M et al (2000) P53 germline mutations in childhood cancers and cancer risk for carrier individuals Br J Cancer 82(12) 1932–1937 PMID: 10864200 PMCID: 2363254
8. Kamihara J, Rana HQ and Garber JE (2014) Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome Hum Mutat 35(6) 654–662 DOI: 10.1002/humu.22559 PMID: 24706533
9. Villani A, Tabori U and Schiffman J et al (2011) Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study Lancet Oncol 12(6) 559–567 DOI: 10.1016/S1470-2045(11)70119-X PMID: 21601526
10. Ruijs MWG, Schmidt MK and Nevanlinna H et al (2007) The single-nucleotide polymorphism 309 in the MDM2 gene contributes to the Li-Fraumeni syndrome and related phenotypes Eur J Hum Genet 15(1) 110–114 DOI: 10.1038/sj.ejhg.5201715
11. Post SM, Quintás-Cardama A and Pant V et al (2010) A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development Cancer Cell 18(3) 220–230 DOI: 10.1016/j.ccr.2010.07.010 PMID: 20832750 PMCID: 2944041
12. Dutt S, Narla A and Lin K et al (2011) Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells Blood 117(9) 2567–2576 DOI: 10.1182/blood-2010-07-295238 PMCID: 3062351
13. Van Nostrand JL, Brady CA and Jung H et al (2014) Inappropriate p53 activation during development induces features of CHARGE syndrome Nature 127(7521) 358–366
14. Lee Y, Shull ER and Frappart PO et al (2012) ATR maintains select progenitors during nervous system development EMBO J 31(5) 1177–1189 DOI: 10.1038/emboj.2011.493 PMID: 22266795 PMCID: 3298000
15. Srivastava S, Zou ZQ and Pirollo K (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome Nature 348(6303) 747–749 DOI: 10.1038/348747a0 PMID: 2259385
16. Rivlin N, Brosh R and Oren M (2011) Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis Genes Cancer 2(4) 466–474 DOI: 10.1177/1947601911408889 PMID: 21779514 PMCID: 3135636
17. Parant JM, George SA and Holden JA et al (2010) Genetic modeling of Li-Fraumeni syndrome in zebrafish Dis Models Mech 3(1–2) 45–56 DOI: 10.1242/dmm.003749
18. Knudson AG (1971) Mutation and cancer: statistical study of retinoblastoma Proc Natl Acad Sci USA 68(4) 820–823 DOI: 10.1073/pnas.68.4.820 PMID: 5279523 PMCID: 389051
19. Heymann S, Delaloge S and Rahal A et al (2010) Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome Radiat Oncol 5(1) 104 DOI: 10.1186/1748-717X-5-104
20. Rufini A, Tucci P and Celardo I et al (2013) Senescence and aging: the critical roles of p53 Oncogene 32(43) 5129–5143 DOI: 10.1038/onc.2012.640 PMID: 23416979
21. Sahin E and De Pinho RA (2012) Axis of ageing: telomeres, p53 and mitochondria Nat Rev MolCell Biol 13(6) 397–404 DOI: 10.1038/nrm3352
22. Wang SJ and Gu W (2014) To be, or not to be: functional dilemma of p53 metabolic regulation Curr Opin Oncol 26(1) 78–85 DOI: 10.1097/CCO.0000000000000024 PMCID: 3968813
23. Cheung EC and Vousden KH (2010) The role of p53 in glucose metabolism Curr Opin Cell Biol 22(2) 186–191 DOI: 10.1016/j.ceb.2009.12.006 PMID: 20061129
24. Zhuang J, Ma W and Lago CU et al (2012) Metabolic regulation of oxygen and redox homeostasis by p53: lessons from evolutionary biology? Free Radical Bio Med 53(6) 1279–1285 DOI: 10.1016/j.freeradbiomed.2012.07.026
25. Menendez D, Shatz M and Resnick MA (2013) Interactions between the tumor suppressor p53 and immune responses Current Opin Oncol 25(1) 85–92 DOI: 10.1097/CCO.0b013e32835b6386
26. Iannello A, Thompson TW and Ardolino M et al (2013) p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells J Exp Med 210(10) 2057–2069 DOI: 10.1084/jem.20130783 PMID: 24043758 PMCID: 3782044
27. Lujambio A, Akkari L and Simon J et al (2013) Non-cell-autonomous tumor suppression by p53 Cell 153(2) 449–460 DOI: 10.1016/j.cell.2013.03.020 PMID: 23562644 PMCID: 3702034
28. Bieging KT, Mello SS and Attardi LD (2014) Unravelling mechanisms of p53-mediated tumour suppression Nat Rev Cancer 14(5) 359–370 DOI: 10.1038/nrc3711 PMID: 24739573 PMCID: 4049238
29. Monti P, Perfumo C and Bisio A et al (2011) Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes Mol Cancer Res 9(3) 271–279 DOI: 10.1158/1541-7786.MCR-10-0496 PMID: 21343334 PMCID: 3077904
30. Garritano S, Inga A and Gemignani F et al (2013) More targets, more pathways and more clues for mutant p53 Oncogenesis 2(7) e54 DOI: 10.1038/oncsis.2013.15 PMID: 23817466 PMCID: 3740285
31. Bisio A, Ciribilli Y and Fronza G et al (2014) TP53 mutants in the tower of babel of cancer progression Hum Mutat 35(6) 689–701 DOI: 10.1002/humu.22514 PMID: 24449472
32. Surget S, Khoury MP and Bourdon JC (2013) Uncovering the role of p53 splice variants in human malignancy: a clinical perspective Onco TargetsTher 7 57–67
33. Senturk S, Yao Z and Camiolo M et al (2014) P53Ψ Is a transcriptionally inactive P53 isoform able to reprogram cells toward a metastatic-like state Proc Natl Acad Sci USA DOI: 10.1073/pnas.1321640111
34. Allen M and Jones LJ (2011) Jekyll and Hyde: the role of the microenvironment on the progression of cancer J Pathol 223(2) 162–176 DOI: 10.1002/path.2803
35. Quail DF and Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis Nat Med 19(11) 1423–1437 DOI: 10.1038/nm.3394 PMID: 24202395 PMCID: 3954707
36. Hanahan D and Coussens LM (2012) Accessories to the Crime: functions of cells recruited to the tumor microenvironment Cancer Cell 21(3) 309–322 DOI: 10.1016/j.ccr.2012.02.022 PMID: 22439926
37. Pienta KJ, McGregor N and Axelrod R et al (2008) Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments Transl Oncol 1(4) 158–164 DOI: 10.1593/tlo.08178 PMID: 19043526 PMCID: 2582164
38. Tian T, Olson S and Whitacre JM (2011) The origins of cancer robustness and evolvability Integr Biol 3(1) L17–30 DOI: 10.1039/C0IB00046A
39. Paget S (1889) The distribution of secondary growths in cancer of the breast Lancet 133(3421) 571–573 DOI: 10.1016/S0140-6736(00)49915-0
40. Coghlin C and Murray GI (2010) Current and emerging concepts in tumour metastasis J Pathol 222(1) 1–15 DOI: 10.1002/path.2727 PMID: 20681009
41. Kaplan RN, Riba RD and Zacharoulis S et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche Nature 438(7069) 820–827 DOI: 10.1038/nature04186 PMID: 16341007 PMCID: 2945882
42. Kaplan RN, Rafii S and Lyden D (2006) Preparing the ‘soil’: the premetastatic niche Cancer Res 66(23) 11089–11093 DOI: 10.1158/0008-5472.CAN-06-2407 PMID: 17145848 PMCID: 2952469
43. Psaila B, Kaplan RN and Port ER et al (2007) Priming the ‘soil’ for breast cancer metastasis: the pre-metastatic niche Breast Dis 26 65–74
44. Hiratsuka S, Watanabe A and Aburatani H et al (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis Nat Cell Biol 8(12) 1369–1375 DOI: 10.1038/ncb1507 PMID: 17128264
45. Elkabets M, Gifford AM and Scheel C et al (2011) Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice J Clin Invest 121(2) 784–799 DOI: 10.1172/JCI43757 PMID: 21266779 PMCID: 3026724
46. Kuznetsov HS, Marsh T and Markens BA et al (2012) Identification of luminal breast cancers that establish a tumor-supportive macroenvironment defined by proangiogenic platelets and bone marrow-derived cells Cancer Discov 2(12) 1150–1165 DOI: 10.1158/2159-8290.CD-12-0216 PMID: 22896036 PMCID: 3517696
47. Oskarsson T, Acharyya S and Zhang XHF et al (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs Nat Med 17(7) 867–874 DOI: 10.1038/nm.2379 PMID: 21706029 PMCID: 4020577
48. Liu S, Jiang M and Zhao Q et al (2014) Vascular endothelial growth factor plays a critical role in the formation of the pre-metastatic niche via prostaglandin E2 Oncol Rep 2477–2484 PMID: 25333935
49. McAllister SS and Weinberg RA (2014) The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis Nat Cell Biol 16(8) 717–727 DOI: 10.1038/ncb3015 PMID: 25082194
50. Ansell SM and Vonderheide RH (2013) Cellular composition of the tumor microenvironment Am Soc Clin Oncol Educ Book Meeting 2013 91–97 DOI: 10.1200/EdBook_AM.2013.33.e91
51. Kareva I (2011) What can ecology teach us about cancer? Transl Oncol 4(5) 266–270 DOI: 10.1593/tlo.11154 PMID: 21966543 PMCID: 3162301
52. Parolini I, Federici C and Raggi C et al (2009) Microenvironmental pH is a key factor for exosome traffic in tumor cells J Biol Chem 284(49) 34211–34222 DOI: 10.1074/jbc.M109.041152 PMID: 19801663 PMCID: 2797191
53. Bernhard EJ (2011) Interventions that induce modifications in the tumor microenvironment Cancer Radiother 15(5) 376–382 DOI: 10.1016/j.canrad.2011.01.007 PMID: 21571567
54. Barcellos-Hoff MH, Lyden D and Wang TC (2013) The evolution of the cancer niche during multistage carcinogenesis Nat Rev Cancer 13(7) 511–518 DOI: 10.1038/nrc3536 PMID: 23760023
55. Bussard KM and Smith GH (2012) Human breast cancer cells are redirected to mammary epithelial cells upon interaction with the regenerating mammary gland microenvironment in-vivo PloS one 7(11) e49221 DOI: 10.1371/journal.pone.0049221 PMID: 23155468 PMCID: 3498340
56. Rosenfield SM and Smith GH (2013) Redirection of human cancer cells upon the Interaction with the regenerating mouse mammary gland microenvironment Cells 2(1) 43–56 DOI: 10.3390/cells2010043 PMID: 24709643 PMCID: 3972660
57. Dezorella N, Pevsner-Fischer M and Deutsch V et al (2009) Mesenchymal stromal cells revert multiple myeloma cells to less differentiated phenotype by the combined activities of adhesive interactions and interleukin-6 Exp Cell Res 315(11) 1904–1913 DOI: 10.1016/j.yexcr.2009.03.016 PMID: 19328780
58. Grivennikov SI, Greten FR and Karin M (2010) Immunity, inflammation, and cancer Cell 140(6) 883–899 DOI: 10.1016/j.cell.2010.01.025 PMID: 20303878 PMCID: 2866629
59. Federico A, Morgillo F and Tuccillo C et al (2007) Chronic inflammation and oxidative stress in human carcinogenesis Int J Cancer 121(11) 2381–2386 DOI: 10.1002/ijc.23192 PMID: 17893868
60. Solinas G, Marchesi F and Garlanda C et al (2010) Inflammation-mediated promotion of invasion and metastasis Cancer Metast Rev 29(2) 243–248 DOI: 10.1007/s10555-010-9227-2
61. Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation Cell 144(5) 646–674 DOI: 10.1016/j.cell.2011.02.013 PMID: 21376230
62. Coussens LM and Werb Z (2010) Inflammation and cancer Nature 420(6917) 860–897 DOI: 10.1038/nature01322 PMCID: 2803035
63. Cooks T, Harris CC and Oren M (2014) Caught in the cross fire: p53 in inflammation Carcinogenesis 35(8) 1680–1690 DOI: 10.1093/carcin/bgu134 PMID: 24942866 PMCID: 4123652
64. Yang L and Karin M (2014) Roles of tumor suppressors in regulating tumor-associated inflammation Cell Death Differ 1–10
65. Schwitalla S, Ziegler PK and Horst D et al (2013) Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors Cancer Cell 23(1) 93–106 DOI: 10.1016/j.ccr.2012.11.014 PMID: 23273920
66. Staib F, Robles AI and Varticovski L et al (2005) The p53 tumor suppressor network is a key responder to microenvironmental components of chronic inflammatory stress Cancer Res 65(22) 10255–10264 DOI: 10.1158/0008-5472.CAN-05-1714 PMID: 16288013 PMCID: 1421332
67. Hussain SP, Amstad P and Raja K et al (2000) Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease Cancer Res 60(13) 3333–3337 PMID: 10910033
68. Cooks T, Pateras IS and Tarcic O et al (2013) Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer Cancer Cell 23(5) 634–646 DOI: 10.1016/j.ccr.2013.03.022 PMID: 23680148 PMCID: 3657134
69. Yamanishi Y, Boyle DL and Pinkoski MJ et al (2002) Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis Am J Pathol 160(1) 123–130 DOI: 10.1016/S0002-9440(10)64356-8 PMID: 11786406 PMCID: 1867134
70. Macedo GS, Lisbôa da Motta L and Giacomazzi J et al (2012) Increased oxidative damage in carriers of the germline TP53 p.R337H mutation PLoS ONE 7(10) e47010 DOI: 10.1371/journal.pone.0047010 PMID: 23056559 PMCID: 3467233
71. West XZ, Malinin NL and Merkulova AA et al (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands Nature 467(7318) 972–976 DOI: 10.1038/nature09421 PMID: 20927103 PMCID: 2990914
72. Kim YW, West XZ and Byzova TV (2013) Inflammation and oxidative stress in angiogenesis and vascular disease J Mol Med 91(3) 323–328 DOI: 10.1007/s00109-013-1007-3 PMID: 23430240 PMCID: 3656485
73. Dameron KM, Volpert OV and Tainsky MA et al (1994) Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1 Science 265(5178) 1582–1584 DOI: 10.1126/science.7521539 PMID: 7521539
74. Su F, Pascal LE and Xiao W et al (2010) Tumor suppressor U19/EAF2 regulates thrombospondin-1 expression via p53 Oncogene 29(3) 421–431 DOI: 10.1038/onc.2009.326 PMCID: 2809809
75. Ioachim E, Damala K and Tsanou E et al (2012) Thrombospondin-1 expression in breast cancer: prognostic significance and association with p53 alterations, tumour angiogenesis and extracellular matrix components Histol Histopathol 27(2) 209–216
76. Grossfeld GD, Ginsberg DA and Stein JP et al (1997) Thrombospondin-1 expression in bladder cancer: association with p53 alterations, tumor angiogenesis, and tumor progression J Natl Cancer Inst 89(3) 219–227 DOI: 10.1093/jnci/89.3.219 PMID: 9017002
77. Hermeking H (2012) MicroRNAs in the p53 network: micromanagement of tumour suppression Nat Rev Cancer 12(9) 613–626 DOI: 10.1038/nrc3318 PMID: 22898542
78. Yamakuchi M, Lotterman CD and Bao C et al (2010) P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis Proc Natl Acad Sci USA 107(14) 6334–6339 DOI: 10.1073/pnas.0911082107 PMID: 20308559 PMCID: 2851979
79. De La Peña FA, Kanasaki K and Kanasaki M et al (2011) Loss of p53 and acquisition of angiogenic microRNA profile are insufficient to facilitate progression of bladder urothelial carcinoma in situ to invasive carcinoma J Biol Chem 286(23) 20778–20787 DOI: 10.1074/jbc.M110.198069
80. Volpert O, Dameron KM and Bouck N (1997) Sequential development of an angiogenic phenotype by human fibroblasts progressing to tumorigenicity Oncogene 14(12) 1495–1502 DOI: 10.1038/sj.onc.1200977 PMID: 9136993
81. Fontemaggi G, Dell’Orso S and Trisciuoglio D et al (2009) The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis Nat Struct Mol Biol 16(10) 1086–1093 DOI: 10.1038/nsmb.1669 PMID: 19783986
82. Xu J, Qian J and Hu Y et al (2014) Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations Sci Rep 4 4223 PMID: 24573247 PMCID: 3936234
83. Huang Y, Yu P and Li W et al (2014) P53 regulates mesenchymal stem cell-mediated tumor suppression in a tumor microenvironment through immune modulation Oncogene 33(29) 3830–3838 DOI: 10.1038/onc.2013.355
84. Shatz M, Menendez D and Resnick MA (2012) The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells Cancer Res 72(16) 3948–3957 DOI: 10.1158/0008-5472.CAN-11-4134 PMID: 22673234 PMCID: 3443562
85. Hoebe K, Janssen E and Beutler B (2004) The interface between innate and adaptive immunity Nat Immunol 5(10) 971–974 DOI: 10.1038/ni1004-971 PMID: 15454919
86. Huang B, Zhao J and Li H et al (2005) Toll-like receptors on tumor cells facilitate evasion of immune surveillance Cancer Res 65(12) 5009–5014 DOI: 10.1158/0008-5472.CAN-05-0784 PMID: 15958541
87. Rakoff-Nahoum S and Medzhitov R (2009) Toll-like receptors and cancer Nat Rev Cancer 9(1) 57–63 DOI: 10.1038/nrc2541
88. Basith S, Manavalan B and Yoo TH et al (2012) Roles of toll-like receptors in cancer: a double-edged sword for defense and offense Arch Pharmacal Res 35(8) 1297–1316 DOI: 10.1007/s12272-012-0802-7
89. Sato Y, Goto Y and Narita N et al (2009) Cancer cells expressing toll-like receptors and the tumor microenvironment Cancer Microenviron 2(Suppl 1) 205–214 DOI: 10.1007/s12307-009-0022-y PMID: 19685283 PMCID: 2756339
90. Menendez D, Shatz M and Azzam K et al (2011) The toll-like receptor gene family is integrated into human DNA damage and p53 networks PLoS genetics 7(3) e1001360 DOI: 10.1371/journal.pgen.1001360 PMID: 21483755 PMCID: 3069118
91. Yao S, Zhu Y and Chen L (2013) Advances in targeting cell surface signalling molecules for immune modulation Nat Rev Drug Discov 12(2) 130–146 DOI: 10.1038/nrd3877 PMID: 23370250 PMCID: 3698571
92. Cortez MA, Valdecanas D and Wang X et al (2015) p53 regulation of PDL1 is mediated through miR-34a Proc AACR 2015 (Abstract 2875)
93. Ferreira LMR, Hebrant A and Dumont JE (2012) Metabolic reprogramming of the tumor Oncogene 31(36) 3999–4011 DOI: 10.1038/onc.2011.576 PMID: 22231450
94. Olovnikov IA, Kravchenko JE and Chumakov PM (2009) Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense Semin Cancer Biol 19(1) 32–41 DOI: 10.1016/j.semcancer.2008.11.005 PMCID: 2646792
95. Bonuccelli G, Whitaker-Menezes D and Castello-Cros R et al (2010) The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts Cell Cycle 9(10) 1960–1971 DOI: 10.4161/cc.9.10.11601 PMID: 20495363
96. Fiaschi T, Marini A and Giannoni E et al (2012) Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay Cancer Res 72(19) 5130–5140 DOI: 10.1158/0008-5472.CAN-12-1949 PMID: 22850421
97. Martinez-Outschoorn UE, Pavlides S and Howell A et al (2011) Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment Int J Biochem Cell Biol 43(7) 1045–1051 DOI: 10.1016/j.biocel.2011.01.023 PMID: 21300172 PMCID: 3102770
98. Witkiewicz AK, Whitaker-Menezes D and Dasgupta A et al (2012) Using the ‘reverse Warburg effect’ to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers Cell Cycle 11(6) 1108–1117 DOI: 10.4161/cc.11.6.19530 PMID: 22313602 PMCID: 3335917
99. Pértega-Gomes N, Vizcaíno JR and Attig J et al (2014) A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer BMC Cancer 14(1) 352 DOI: 10.1186/1471-2407-14-352 PMID: 24886074 PMCID: 4039335
100. Sotgia F, Martinez-Outschoorn UE and Lisanti MP (2014) The reverse warburg effect in osteosarcoma Oncotarget 5(18) 7982–7983 PMID: 25478627 PMCID: 4226660
101. Pantziarka P (2013) Li Fraumeni syndrome, cancer and senescence: a new hypothesis Cancer Cell Int 13(1) 35 DOI: 10.1186/1475-2867-13-35 PMID: 23587008 PMCID: 3637189
102. Mercier I, Camacho J and Titchen K et al (2012) Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug Am J Pathol 181(1) 278–293 DOI: 10.1016/j.ajpath.2012.03.017 PMID: 22698676 PMCID: 3388151
103. Sherif ZA and Sultan AS (2013) Divergent control of Cav-1 expression in non-cancerous Li-Fraumeni syndrome and human cancer cell lines Cancer Biol Ther 14(1) 29–38 DOI: 10.4161/cbt.22621 PMCID: 3566049
104. Wang X, Docanto MM and Sasano H et al (2015) Prostaglandin E2 inhibits p53 in human breast adipose stromal cells : a novel mechanism for the regulation of aromatase in obesity and breast cancer Cancer Res 2 1–12
105. Trkova M, Prochazkova K and Krutilkova V (2007) Telomere length in peripheral blood cells of germline TP53 mutation carriers is shorter than that of normal individuals of corresponding age Cancer 110(3) 694–702 DOI: 10.1002/cncr.22834 PMID: 17567834
106. Kruk PA and Bohr VA (1999) Telomeric length in individuals and cell lines with altered p53 status Radiat Oncol Investig 7(1) 13–21 PMID: 10030619
107. Pinto C, Veiga I and Pinheiro M et al (2009) TP53 germline mutations in Portugal and genetic modifiers of age at cancer onset Fam Cancer 8(4) 383–390 DOI: 10.1007/s10689-009-9251-y PMID: 19468865
108. Tabori U, Nanda S and Druker H et al (2007) Younger age of cancer initiation is associated with shorter telomere length in Li-Fraumeni syndrome Cancer Res 67(4) 1415–1418 DOI: 10.1158/0008-5472.CAN-06-3682 PMID: 17308077
109. Boyle JM, Mitchell EL and Greaves MJ et al (1998) Chromosome instability is a predominant trait of fibroblasts from Li-Fraumeni families Br J Cancer 77(12) 2181–2192 DOI: 10.1038/bjc.1998.364 PMID: 9649131 PMCID: 2150396
110. Bischoff FZ, Yim SO and Pathak S et al (1990) Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: aneuploidy and immortalization Cancer Res 50(24) 7979–7984 PMID: 2253239
111. Shay JW, Tomlinson G and Piatyszek MA et al (1995) Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome Mol Cell Biol 15(1) 425–432 PMID: 7799951 PMCID: 231985
112. Tazawa H, Tatemichi M and Sawa T et al (2007) Oxidative and nitrative stress caused by subcutaneous implantation of a foreign body accelerates sarcoma development in Trp53 /- mice Carcinogenesis 28(1) 191–198 DOI: 10.1093/carcin/bgl128
113. Komarova EA, Antoch MP and Novototskaya LR et al (2012) Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53 /- mice Aging 4(10) 709–714 PMID: 23123616 PMCID: 3517941
114. Berrigan D, Perkins SN and Haines DC et al (2002) Adult-onset calorie restriction and fasting delay spontaneous tumorigenesis in p53-deficient mice Carcinogenesis 23(5) 817–822 DOI: 10.1093/carcin/23.5.817 PMID: 12016155
115. Eikawa S, Nishida M and Mizukami S et al (2015) Immune-mediated antitumor effect by type 2 diabetes drug, metformin Proc Natl Acad Sci USA 1–6
116. Pantziarka P, Bouche G and Meheus L et al (2014) The repurposing drugs in oncology (ReDO) project Ecancermedicalscience 8 442 DOI: 10.3332/ecancer.2014.485 PMID: 25075216 PMCID: 4096030