Cancer stem cells and post-therapy tumour recurrence: a systematic review of mechanistic pathways and translational gaps
Imad Barjij1,2,a and Meryem Meliani3
1Medical Oncology Department, National Institute of Oncology, Ibn Sina University Hospital, Rabat 10000, Morocco
2Faculty of Medicine and Pharmacy of Rabat, Mohammed V University of Rabat, Rabat 10000, Morocco
3Medical Oncology Department, Regional Oncology Center of Laâyoune, Regional Hospital Center of Laâyoune, Laâyoune 70000, Morocco
ahttps://orcid.org/0009-0004-2172-1058
Abstract
Background: Cancer stem cells (CSCs) are increasingly recognised as pivotal drivers of tumour recurrence and treatment resistance across multiple malignancies. Despite extensive preclinical investigations, the mechanisms by which CSCs mediate relapse after therapy remain insufficiently integrated and poorly translated into clinical frameworks.
Objective: This systematic review aimed to synthesise current mechanistic evidence linking CSC biology to post-therapeutic recurrence in solid and hematologic tumours, highlighting recurrent molecular pathways, experimental models and translational gaps.
Methods: Following Preferred Reporting Items for Systematic Reviews and Meta-Analyses 2020 guidelines, five major databases (PubMed, Scopus, Web of Science, Embase and ResearchRabbit) were searched without time or language restriction. Eligible studies included original experimental articles investigating the mechanistic role of CSCs in tumour recurrence, therapy resistance or metastatic relapse. A total of 23 studies were included after rigorous screening and data extraction. Risk of bias was assessed using predefined methodological criteria. No meta-analysis was conducted due to mechanistic and qualitative heterogeneity.
Results: Included studies spanned diverse tumour types, including glioblastoma, breast, pancreatic, hepatocellular, colorectal, lung, thyroid and hematologic cancers. CSC-related recurrence was linked to key mechanistic axes: chromatin remodeling (e.g., suppressor of variegation 3-9 Homolog 1, methyltransferase like 16), transcriptional regulators (e.g., SRY-box transcription factor 2, MYC and Transcription factor activating enhancer-binding protein 4), epithelial-to-mesenchymal transition-associated plasticity, immune evasion (Programmed death-ligand 1, stimulator of interferon genes pathway suppression), metabolic rewiring (P-element induced WImpy Testis-like 2–PDK1, ribosomal biosynthesis) and microenvironmental crosstalk (cancer-associated fibroblasts- and myeloid-derived suppressor cells-mediated niches). Across studies, CSCs demonstrated higher resistance to chemotherapy, prolonged survival under treatment stress and robust capacity for tumour regeneration.
Limitations: The majority of studies were preclinical and varied in CSC definitions and recurrence models. Few incorporated longitudinal tracking or patient-level validation. Overall risk of bias was moderate due to lack of blinding, protocol registration or replication.
Conclusion: CSC-driven recurrence is a multifaceted and dynamic process shaped by epigenetic, transcriptional, metabolic and immunologic adaptations. Single-target strategies are unlikely to achieve durable eradication. Future research must prioritise multi-targeted approaches, integrate CSC endpoints into clinical trials and develop predictive biomarkers of CSC burden. Addressing CSC-mediated relapse is essential for advancing precision oncology and achieving lasting therapeutic responses.
Keywords: cancer stem cells, recurrence, therapy resistance, tumour relapse, epigenetics, EMT, tumour microenvironment, immunoevasion, mechanistic pathways, systematic review
Correspondence to: Imad Barjij
Email: imad_barjij@um5.ac.ma
Published: 17/10/2025
Received: 16/05/2025
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Background and rationale
Tumour recurrence following initial response to therapy remains one of the most critical challenges in oncology today. Despite significant advances in targeted therapies, immunotherapy and precision medicine, a substantial proportion of patients experience relapse, often more aggressive and treatment-resistant than the primary disease. Over the past two decades, a growing body of evidence has attributed this phenomenon, at least in part, to the persistence and adaptability of cancer stem cells (CSCs), a subpopulation of tumour cells endowed with self-renewal capacity, multipotency and resistance to conventional cytotoxic agents [1–7].
CSCs have now been identified in a wide spectrum of malignancies, including glioblastoma, breast cancer, hepatocellular carcinoma (HCC), colorectal cancer, non-small cell lung cancer (NSCLC), cervical cancer, ovarian carcinoma, pancreatic adenocarcinoma and hematologic cancers such as acute myeloid leukaemia (AML). These cells are not only capable of initiating tumours but are also implicated in key processes that drive therapeutic resistance, tumour dormancy, epithelial-to-mesenchymal transition (EMT), immune escape, metabolic reprogramming and eventual relapse. Functionally, CSCs are characterised by specific surface markers (e.g., cluster of differentiation (CD)44, CD133, Aldehyde Dehydrogenase (ALDH1)), enhanced DNA repair capacity, epigenetic plasticity and a dynamic interaction with the tumour microenvironment (TME), including immune cells and stromal components [1, 5, 8–11].
While the conceptual framework of CSCs has been extensively explored, the precise mechanisms by which these cells evade treatment and regenerate tumours remain incompletely understood. Existing reviews have provided valuable insights into selected molecular pathways, such as Wnt/β-catenin, Hedgehog, Notch and PI3K/AKT, but often focus on individual tumour types or therapeutic classes. Few have attempted to systematically integrate experimental evidence across multiple cancer types, encompassing diverse mechanistic insights into how CSCs contribute to post-treatment recurrence [1, 2, 5, 7, 12].
Furthermore, the translational potential of CSC-targeting strategies is still limited by a lack of mechanistic clarity. Despite promising preclinical studies, efforts to eliminate CSCs or prevent their reactivation post-therapy have yet to yield broadly effective clinical applications. Contributing to this gap is the fragmented nature of existing data, often scattered across isolated in vitro studies or small-scale in vivo models, without comprehensive synthesis [5, 10, 13–15].
Given this context, there is a clear need for a structured and critical appraisal of original experimental research exploring the role of CSCs in post-therapy tumour recurrence. Such a synthesis is essential not only to identify conserved molecular themes and vulnerabilities but also to highlight evidence gaps and inform the rational development of CSC-directed therapeutic interventions. This is particularly relevant as emerging combination strategies, such as immunotherapy paired with CSC-targeting agents, gain traction in translational oncology [4, 6, 8, 10, 13, 16].
This review, therefore, aims to address this unmet need by systematically evaluating and integrating experimental studies that investigate CSCs in the context of therapeutic relapse, resistance and disease progression. Unlike narrative reviews or meta-analyses limited to clinical outcomes, this work focuses specifically on mechanistic pathways and experimental evidence, spanning in vitro assays, in vivo models (including patient-derived xenografts (PDXs)), organoid systems and omics-based approaches. In doing so, it provides a detailed landscape of
CSC-driven recurrence mechanisms, identifies common regulatory networks and proposes translational directions grounded in experimental biology.
Objectives
This systematic review aims to critically synthesise experimental evidence on the role of CSCs in post-therapy tumour recurrence, with an emphasis on mechanistic pathways and biological processes implicated in relapse, resistance and disease progression. Specifically, the review addresses the following objectives:
- To identify and summarise original experimental studies (in vitro, in vivo, ex vivo or multi-omics) that explore the contribution of CSCs to tumour recurrence following treatment.
- To classify and analyse the underlying molecular and cellular mechanisms enabling CSC survival, maintenance and expansion after therapy.
- To highlight key evidence gaps and limitations in the current mechanistic literature and provide recommendations for future translational or therapeutic strategies.
This review follows the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 reporting standards and is based on a rigorously curated set of 23 original studies (Table 1), selected according to strict inclusion criteria focused on CSC identification, experimental validity and relevance to therapeutic recurrence. To the best of our knowledge, this is the first systematic synthesis that bridges experimental oncology and stem cell biology to comprehensively map CSC-driven relapse mechanisms across diverse tumour entities.
Methods
Eligibility criteria
We defined clear and stringent eligibility criteria to ensure the inclusion of original experimental studies with high mechanistic value related to CSCs and post-therapy tumour recurrence. Eligible studies had to meet the following criteria:
- Study type: Only original experimental studies were included. This encompassed in vitro studies, in vivo animal models (e.g., xenografts, syngeneic or transgenic models), organoid models, PDXs and multi-omics investigations. Studies combining these methods were prioritised.
- Cancer type: All solid tumours and hematologic malignancies were considered eligible if CSCs were explicitly investigated in the context of therapeutic relapse or recurrence.
- CSC focus: Studies must have clearly identified CSCs or tumour-initiating cells (TICs) using established markers (e.g., CD44, CD133, ALDH1, extramedullary infiltration (EpCAM)) or functional assays (e.g., tumoursphere formation and extreme limiting dilution assay (ELDA)). Studies lacking specific CSC markers or mechanistic data were excluded.
- Outcome of interest: Only studies assessing recurrence, resistance or survival advantage of CSCs post-therapy were included. Studies focused solely on tumourigenesis without any link to therapy or relapse were excluded.
- Publication characteristics: Only full-text, peer-reviewed articles in English were included. Reviews, editorials, commentaries, conference abstracts and study protocols were excluded. Time frame: No date restriction was applied to the initial search. While the majority of eligible studies were published between 2003 and 2024, the review also included studies published as recently as 2025, enhancing the recency and relevance of the synthesis.
Included studies were grouped and synthesised according to cancer type and mechanistic pathway (e.g., Wnt/β-catenin, EMT and immune evasion), as reflected in Table 1.
Table 1. Summary of included studies and key findings.
Information sources
To identify relevant studies, we searched the following bibliographic databases:
- PubMed/MEDLINE (via NCBI)
- Scopus (Elsevier)
- Web of Science Core Collection (Clarivate Analytics)
- Embase (Elsevier)
- Google Scholar (manual selection from first 200 hits)
All databases were searched between 15 January and 10 February 2025.
Additionally, we manually screened the reference lists of included articles and relevant systematic reviews to identify studies not captured through database searches.
No grey literature, trial registries, unpublished data or non-English articles were included.
Search strategy
Search strategies combined MeSH terms and free-text keywords. An example of the PubMed strategy is as follows:
(‘CSC’ OR ‘TIC’ OR ‘TIC’ OR ‘CSCs’ OR ‘TICs’) AND (‘recurrence’ OR ‘relapse’ OR ‘resistance’ OR ‘residual disease’ OR ‘post-therapy’) AND (‘mechanism’ OR ‘pathway’ OR ‘signaling’ OR ‘molecular’) AND (‘in vitro’ OR ‘in vivo’ OR ‘xenograft’ OR ‘PDX’ OR ‘organoid’ OR ‘transcriptomic’ OR ‘multi-omics’).
We applied filters for English language and peer-reviewed journal articles. No restriction on publication date was imposed.
Selection process
Two reviewers (I.B. and M.M.) independently screened titles and abstracts. Full texts were retrieved for all studies deemed potentially eligible. Disagreements were resolved through discussion between the two reviewers.
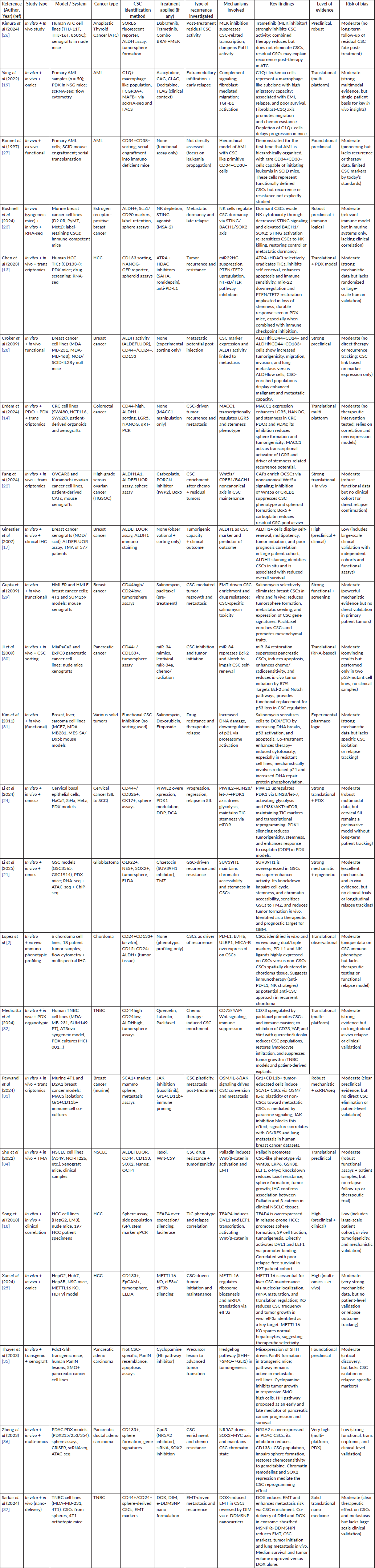
The study selection process is illustrated in Figure 1 below. Of the 1,148 records initially identified, 985 were screened after removal of duplicates and non-eligible records. Following full-text assessment of 163 reports, a total of 23 studies met the predefined inclusion criteria and were incorporated into the final qualitative synthesis.
Data collection process
Two reviewers independently extracted data from each included study using a pre-designed template. The data collection focused on study design, model/system used, cancer type, CSC identification method, treatment applied, type of recurrence investigated, molecular mechanisms described and key outcomes. Any disagreements were resolved through consensus.
No automation tools were used. When information was missing or unclear, supplementary materials or full-text reanalysis were consulted. No authors were contacted.
Data items
Outcomes
The primary outcome was the mechanistic role of CSCs in post-therapy tumour recurrence, including: - Tumour regrowth or resistance after chemotherapy, radiotherapy or targeted therapy - CSC enrichment or maintenance following treatment - Identification of signaling pathways or epigenetic changes driving CSC-mediated relapse.
Secondary outcomes included: - Immune evasion mechanisms - Metastatic dormancy and late relapse - Therapeutic response modulation via CSC pathways.
Other variables
Other variables extracted included:
- Cancer type and subtype
- Model system (cell lines, organoids and animal models)
- Type and duration of treatment applied
- Functional assays used to define CSC activity
- Omics techniques (e.g., RNA sequencing (RNA-seq), ATAC-seq and chromatin immunoprecipitation sequencing (ChIP-seq))
When data were missing (e.g., duration of follow-up or expression levels), we documented the lack transparently without imputation.
Risk of bias assessment
Risk of bias refers to the potential for systematic errors that could distort the interpretation of a study’s findings. In this review, the risk of bias was assessed for each study by two reviewers based on methodological rigor:
- Type of model used (e.g., PDX versus cell line)
- Clarity and reproducibility of CSC identification
- Presence of functional validation (e.g., limiting dilution assays)
- Relevance to recurrence or resistance
No standardised risk-of-bias tool exists for mechanistic CSC studies; we therefore developed a qualitative judgment scale with three levels: Low, moderate and high, justified in Table 1. Disagreements in assessment were resolved by consensus.
Effect measures
No quantitative synthesis or meta-analysis was planned, as this review focused on mechanistic pathways rather than effect sizes. As such, no statistical effect measures (e.g., odds ratios and hazard ratios) were applied. A meta-analysis was deemed methodologically inappropriate due to the heterogeneity of experimental designs, model systems, outcome definitions and mechanistic endpoints across the included studies.
Instead, outcomes were reported narratively and synthesised through thematic classification of biological mechanisms (e.g., Wnt activation and metabolic reprogramming).
Synthesis methods
We tabulated study characteristics and outcomes in Table 1. Studies were grouped according to:
- Cancer type (e.g., glioblastoma, breast cancer and AML)
- Experimental system (e.g., in vitro, in vivo and multi-omics)
- Pathway or mechanism (e.g., EMT, immune escape and chromatin regulation)
This classification enabled a structured narrative synthesis highlighting converging mechanisms and evidence gaps.
We did not conduct sensitivity analyses or subgroup stratifications due to the descriptive nature of the synthesis.
Reporting bias and certainty assessment
We did not conduct formal assessments of publication bias or certainty of evidence (e.g., GRADE), as this review is not focused on intervention outcomes or effect estimation.
However, we qualitatively discuss potential gaps in the literature, including overrepresentation of certain cancer types and underreporting of immune-based CSC mechanisms.
Results
Study selection
A total of 23 original studies were selected based on the predefined eligibility criteria. Figure 1 provides a detailed summary of the selection process in accordance with PRISMA 2020 guidelines.z
Figure 1. PRISMA flow diagram.
Characteristics of included studies
The 23 included studies span publication years from 1997 to 2025, with a sharp increase in publications from 2022 onward and a notable concentration of high-quality mechanistic studies published in 2024 and 2025. The included articles cover a broad spectrum of solid and hematologic malignancies, including glioblastoma, pancreatic ductal adenocarcinoma, AML, breast cancer, HCC, NSCLC, colorectal cancer, triple-negative breast cancer (TNBC), ovarian cancer and chordoma.
Most studies used multi-platform designs, integrating in vitro, in vivo and omics approaches. CSCs were identified using canonical markers (e.g., CD44, CD133 and ALDH1), functional assays (e.g., tumoursphere formation, label-retention and ELDA) and transcriptomic signatures. Several studies employed xenograft or patient-derived models (PDX), while others incorporated clinical correlation data, notably those by Ginestier et al [17], Song et al [18] and Yang et al [19].
Risk of bias
Risk of bias was assessed qualitatively based on study design, mechanistic rigor, translational relevance and completeness of outcome reporting. The majority of studies were classified as having moderate risk of bias, primarily due to lack of longitudinal patient follow-up, absence of blinded outcome assessments or reliance on single model systems. Only three studies were rated as low risk, all of which included multi-level validation, large patient cohorts or integration of functional assays with clinical data. A justification for each risk rating is provided in Table 1, alongside key mechanistic findings.
Summary of key mechanistic themes driving post-therapy CSC persistence and recurrence
Several recurrent mechanisms and biological themes emerged from the 23 included studies:
- Chromatin remodeling and epigenetic plasticity: Li et al [21] demonstrated that chromatin regulators such as suppressor of variegation 3-9 Homolog 1 (SUV39H1) and NR5A2 maintain CSC stemness and mediate therapy resistance through altered enhancer landscapes and transcriptional plasticity.
- Non-canonical signaling and immune evasion: TME-driven pathways, such as Wnt5a–CREB1–BACH1, programmed death-ligand 1 (PD-L1) upregulation and stimulator of interferon genes (STING) pathway suppression, e.g., [2, 22, 23], were shown to enhance CSC survival post-therapy by impairing immune surveillance and promoting niche-mediated dormancy.
- Metabolic reprogramming: Several studies linked CSC survival to enhanced glycolysis, mitochondrial adaptation and ribosome biogenesis, e.g., [24, 25], often driven by upstream regulators such as P-element induced WImpy Testis-like 2 (PIWIL2), Mammalian target of rapamycin (mTOR) and Methyltransferase Like 16 (METTL16).
- CSC plasticity and reprogramming: Studies such as Peyvandi et al [33] and Sarkar et al [37] highlighted CSC interconversion and plasticity, particularly in response to inflammatory or chemotherapeutic cues, emphasising that CSCs may re-emerge from non-stem-like populations under stress.
- Apoptotic and differentiation escape: Modulators of apoptosis (e.g., miR-34, Notch and Bcl-2) and epigenetic repressors (HDAC and EZH2) were recurrently implicated in preventing therapy-induced cell death or promoting incomplete differentiation.
Heterogeneity across cancer types
The included studies reflect considerable heterogeneity in terms of cancer type, CSC identification method and experimental system. While some mechanisms, such as Wnt signaling, ALDH1-associated resistance and immune escape ligands, were consistently reported across several cancers, others appeared tumour-specific, such as NR5A2-driven recurrence in PDAC or CD73-associated immune evasion in TNBC.
To facilitate comparison, full study characteristics and extracted data are presented in Table 1.
Excluded studies
Among the 139 full-text articles excluded, the majority were non-original reports such as reviews, editorials or commentaries (n = 52). Forty-three studies lacked investigation of cancer stem cells or failed to provide mechanistic data related to recurrence. Twenty-six reports explored preclinical models unrelated to relapse or resistance, and 18 articles were excluded for being protocols or reports without experimental data. Excluded studies were documented and archived for transparency.
Certainty of evidence
While the majority of included studies demonstrate high internal validity and mechanistic plausibility, the certainty of evidence remains moderate overall, owing to limited patient-level outcome data, absence of clinical trials and heterogeneous CSC markers. Studies integrating functional validation, PDXs and patient correlation, e.g., [17, 18, 36] contributed most to the certainty assessment.
Discussion
General interpretation of findings
This systematic review synthesised mechanistic evidence from 23 original studies exploring the role of CSCs in mediating tumour recurrence across a wide spectrum of solid and hematologic malignancies. Despite significant heterogeneity in study designs, cancer types and experimental models, several overarching mechanistic themes emerged. These included transcriptional plasticity, chromatin remodeling, epigenetic regulation, immunoevasion, metabolic reprogramming and microenvironmental interactions. Collectively, the data converge toward a unified concept: CSCs persist through and beyond therapy, driving relapse through dynamic adaptation mechanisms and evasion of cytotoxic pressures [1, 3, 5–7, 38].
Across multiple tumour models (glioblastoma, breast, pancreatic, colorectal, hepatocellular, lung, cervical and thyroid carcinomas), studies consistently reported that CSC-enriched subpopulations demonstrate higher resistance to conventional therapies, enhanced self-renewal capacity and increased metastatic potential. This is exemplified by studies such as Zheng et al [36], where inhibition of specific chromatin regulators (SUV39H1 and NR5A2, respectively) restored chemosensitivity in resistant CSC compartments. Similarly, Gupta et al [29] and Mediratta et al [32] showed that targeting EMT-related pathways or metabolic axes significantly abrogated CSC-driven tumour regrowth and metastasis [9, 10, 14, 20, 21, 33, 37, 39, 40].
These results underscore a critical therapeutic paradox: while bulk tumour cells may respond to treatment, CSCs survive and re-initiate tumours through latent and resistant states. Dormancy and reactivation mechanisms, as illustrated by Bushnell et al [23], suggest a temporally regulated escape from immune surveillance mediated by reduced STING signaling and transcriptional rewiring. Other studies implicated the TME in CSC maintenance, such as Fang et al [22] and Peyvandi et al [33], demonstrating that cancer-associated fibroblasts (CAFs) and myeloid-derived suppressor cells create a niche that supports CSC survival and plasticity [4, 9, 10, 23, 33, 41].
Conceptual synthesis: major mechanistic pathways
Based on the aggregated evidence, we propose a conceptual framework integrating the main pathways underlying CSC-mediated recurrence (Figure 2). These include: [2, 9, 10, 14, 15, 19, 24, 39, 42, 43]
- Epigenetic regulators (e.g., SUV39H1, METTL16) that maintain an open chromatin state for stemness gene expression.
- Transcriptional programs (e.g., SRY-box transcription factor 2 (SOX2), MYC, transcription factor activating enhancer-binding protein 4 (TFAP4)) that govern self-renewal and dedifferentiation.
- EMT and mesenchymal transition pathways that promote invasiveness and chemoresistance.
- Immune evasion mechanisms, including upregulation of PD-L1 and loss of immunogenicity.
- Metabolic rewiring, such as glycolysis activation (via PIWIL2–PDK1 axis) and ribosomal biogenesis.
- TME-driven reinforcement, particularly from CAFs and tumour-educated immune cells.
These pathways are not mutually exclusive; rather, they act in a networked and sometimes redundant fashion to ensure CSC survival. As shown by several studies [18, 21, 34], single-pathway inhibition rarely achieves durable eradication of CSCs, thus supporting the need for multi-targeted strategies [4, 14, 15].
Limitations of the evidence base
Although mechanistically rich, the evidence included in this review is subject to several limitations. First, the majority of studies were preclinical, relying on cell lines, PDXs or mouse models. While these systems offer experimental tractability, they may not fully recapitulate human tumour heterogeneity or immune contexture. Only a minority of studies [17, 18] incorporated substantial patient cohort validation, which limits the generalisability of many findings [13, 17, 18, 44–48].
Second, definitions of CSCs varied considerably across studies. Some relied on phenotypic markers (e.g., CD44+/CD24– and ALDHhigh), others on functional assays (e.g., tumoursphere formation and label retention) and yet others used transcriptomic signatures. This lack of standardisation complicates direct comparison and synthesis. Moreover, recurrence itself was operationalised heterogeneously, ranging from in vivo tumour regrowth post-treatment, to resistance to chemotherapy, to expression of relapse-associated gene programs [9, 13, 15, 39, 49].
Third, few studies have conducted longitudinal assessments of CSC dynamics during and after therapy. Most findings were derived from endpoint analyses rather than temporal tracking. Consequently, we lack a detailed understanding of how CSC populations evolve under treatment pressure, an essential aspect for designing effective eradication strategies [13, 19, 44, 48, 50].
Lastly, the risk of bias in included studies was predominantly moderate, largely due to the absence of blinding, replication or standardised reporting of negative results. None of the studies employed registered protocols and reporting completeness varied.
Limitations of the review process
This review has some intrinsic limitations despite rigorous adherence to PRISMA 2020 guidelines. First, although our search strategy was comprehensive (across five major databases), gray literature and unpublished studies were not included, possibly leading to publication bias. Second, screening, data extraction and bias assessment were conducted by two independent reviewers (I.B. and M.M.), with any disagreements resolved through discussion. While this approach is robust, the absence of a third adjudicator may introduce subjectivity in borderline inclusion decisions [6, 15, 16, 41, 48, 51].
Additionally, no meta-analysis was performed, as the review focused on mechanistic evidence, which is inherently qualitative and heterogenous. Although this limits quantitative synthesis, the narrative integration provided a detailed and translationally meaningful overview of biological pathways [26, 33, 36, 41, 52].
Finally, while the review includes studies up to 2025, reflecting high contemporaneity, it remains a snapshot and does not incorporate living review updates.
Implications for practice, policy and future research
This synthesis has several important implications:
- Therapeutic targeting: CSCs are clearly central to recurrence. However, monotherapies against individual pathways (e.g., Bcl-2 inhibition and Wnt blockade) are unlikely to suffice. Rational combinatorial therapies, targeting epigenetic regulators, metabolic pathways and immune checkpoints, should be prioritised [2, 4, 5, 10, 15].
- Biomarker development: Current CSC markers are heterogeneous and context-dependent. Multi-parametric approaches combining surface markers, transcriptomic profiles and functional assays will be essential to reliably identify CSCs and monitor treatment response [8, 10, 13, 15, 47].
- Clinical trial design: Many clinical trials fail to consider CSC dynamics. Future trials should include CSC burden as an endpoint and utilise longitudinal biopsies or liquid biopsies to assess CSC persistence [2, 10, 13, 47, 50].
- Immunotherapy: The role of CSCs in immune evasion (via PD-L1, natural killer (NK) ligand modulation and dormancy) suggests that CSC-targeted immunotherapies, either via CSC vaccines or NK-based approaches, could augment existing checkpoint inhibitors [4, 5, 8, 10, 23, 32, 33].
- Preclinical modeling: Investment in organotypic cultures, humanised mouse models and time-resolved single-cell approaches is critical to bridge the gap between discovery and clinical translation [45, 53, 54].
In conclusion, this systematic review provides a comprehensive synthesis of 23 original studies elucidating the mechanistic roles of CSCs in cancer recurrence. CSC-mediated relapse involves a complex interplay of genetic, epigenetic, immune and microenvironmental factors. Future therapeutic and diagnostic advances must embrace this complexity and integrate CSC biology into mainstream oncologic paradigms.
Figure 2 below provides a conceptual visual summary of the major pathways involved in CSC-mediated recurrence and highlights the critical knowledge gaps that remain.
Although the development of CSC-targeted strategies has been extensively explored in preclinical settings, translation into clinical trials remains limited. A few early-phase clinical trials have attempted to target CSC-specific pathways or markers. For example, the use of demcizumab (anti-DLL4 monoclonal antibody) in pancreatic and NSCLC showed some initial promise [20], although subsequent trials were terminated due to toxicity and limited efficacy. Another example includes BBI608 (napabucasin), a STAT3 inhibitor investigated in several trials [55, 56], which aimed to target CSC-associated signaling. However, many of these efforts failed to demonstrate consistent clinical benefits. As of today, no randomised clinical trial has definitively validated a CSC-specific therapy as a standard of care. This underscores the persistent gap between mechanistic insights and therapeutic translation.
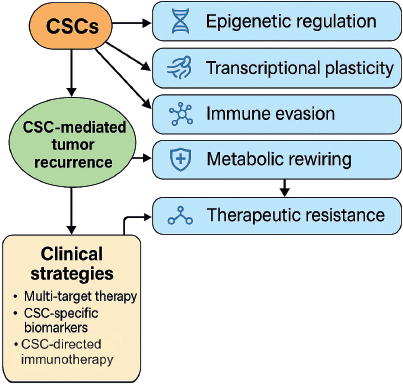
Figure 2. Conceptual diagram of CSC-mediated recurrence mechanisms and biological complexity. Conceptual representation of the major biological mechanisms contributing to CSC-mediated recurrence. The diagram illustrates how epigenetic regulation, transcriptional plasticity, immune evasion, metabolic rewiring and TME-driven reinforcement interact to sustain CSC persistence and therapeutic resistance. The diversity and interplay of these mechanisms underscore the intrinsic biological complexity of CSC-mediated relapse.
Conclusion
CSCs are central drivers of tumour recurrence and therapeutic resistance across diverse cancer types. Despite heterogeneous models, recurrent mechanisms such as chromatin remodeling, immune evasion and metabolic plasticity consistently underlie CSC-mediated relapse. Effective cancer control must integrate CSC-targeted strategies, including immunotherapeutic approaches and biomarker-guided treatments. Recognising CSCs as pivotal entities in recurrence is essential for advancing toward durable remission and curative therapies.
List of abbreviations
ALDH, Aldehyde dehydrogenase; AML, Acute myeloid leukaemia; ATC, Anaplastic thyroid cancer; CAF, Cancer-associated fibroblast; CD, Cluster of differentiation; ChIP-seq, Chromatin immunoprecipitation sequencing; CSC, Cancer stem cell; DDP, Cisplatin (Diamminedichloroplatinum); DIM, 3,3’-Diindolylmethane; DOX, Doxorubicin; e-DDMSNP, Exosome-coated dendritic mesoporous silica nanoparticle; ELDA, Extreme limiting dilution assay; EMI, Extramedullary infiltration; EMT, Epithelial-to-mesenchymal transition; EpCAM, Epithelial cell adhesion molecule; FACS, Fluorescence-activated cell sorting; GSC, Glioblastoma stem cell; HCC, Hepatocellular carcinoma; HDACi, Histone deacetylase inhibitor; IHC, Immunohistochemistry; KO, Knockout; METTL16, Methyltransferase like 16; mTOR, Mammalian target of rapamycin; NK, Natural killer (cell); NSCLC, Non-small cell lung cancer; PD-L1, Programmed death-Ligand 1; PDO, Patient-derived organoid; PDX, Patient-derived xenograft; PIWIL2, P-element induced WImpy Testis-like 2; PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses; qRT-PCR, Quantitative real-time polymerase chain reaction; RNA-seq ,RNA sequencing; SCC, Squamous cell carcinoma; scRNA-seq, Single-cell RNA sequencing; SOX2, SRY-box transcription factor 2; SP, Side population; STING, Stimulator of interferon genes; SUV39H1, Suppressor of variegation 3-9 Homolog 1; TFAP4, Transcription factor activating enhancer-binding protein 4; TIC, Tumour-initiating cell; TMA, Tissue microarray; TME, Tumour microenvironment; TMZ, Temozolomide; ULBP1, UL16-Binding Protein 1; Wnt-C59, Porcupine inhibitor targeting Wnt signaling.
Conflicts of interest
The authors declare no conflicts of interest related to the content of this systematic review. No financial or personal relationships influenced the design, analysis or interpretation of the results.
Funding
This study received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. It was conducted as part of the academic and scientific engagement of the authors.
Ethical approval
As this work is a systematic review of published data, no ethical approval or informed consent was required. All included studies were already approved by their respective institutional review boards.
Data availability
All data generated or analysed during this systematic review are either included in the main article. The complete data extraction table (Table 1), PRISMA flow diagram (Figure 1) and conceptual synthesis figure (Figure 2) are available within the manuscript. Additional information can be obtained from the corresponding author upon request.
References
1. Lee MY, Giraddi RR, and Tam WL (2019) Cancer stem cells: concepts, challenges, and opportunities for cancer therapy Methods Mol Biol https://doi.org/10.1007/978-1-4939-9524-0_4
2. Lopez D, Fabian K, and Padget M, et al (2024) Chordoma cancer stem cell subpopulation characterization may guide targeted immunotherapy approaches to reduce disease recurrence Front Oncol https://doi.org/10.3389/fonc.2024.1376622
3. Nguyen LV, Vanner R, and Dirks PB, et al (2012) Cancer stem cells: an evolving concept Nat RevCancer https://doi.org/10.1038/nrc3184
4. Shibata M and Hoque MO (2019) Targeting cancer stem cells: a strategy for effective eradication of cancer Cancers https://doi.org/10.3390/cancers11050732 PMCID: 6562442
5. Trevisan RLB, Bighetti-Trevisan RL, and Bighetti-Trevisan RL, et al (2019) Cancer stem cells: powerful targets to improve current anticancer therapeutics Stem Cells Int https://doi.org/10.1155/2019/9618065
6. Wang T, Shigdar S, and Gantier MP, et al (2015) Cancer stem cell targeted therapy: progress amid controversies Oncotarget https://doi.org/10.18632/oncotarget.6176
7. Wieczorek K and Niewiarowska J (2012) Cancer stem cells Postȩpy Hig Med Dośw https://doi.org/10.5604/17322693.1009706
8. Davies S, Beckenkamp A, and Buffon A (2015) CD26 a cancer stem cell marker and therapeutic target Biomed Pharmacother https://doi.org/10.1016/j.biopha.2015.02.031 PMID: 25960228
9. Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation Cell https://doi.org/10.1016/j.cell.2011.02.013 PMID: 21376230
10. Huang R and Rofstad EK (2017) Cancer stem cells (CSCs), cervical CSCs and targeted therapies Oncotarget https://doi.org/10.18632/oncotarget.10169
11. Luo M, Brooks M, and Brooks MD, et al (2015) Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance Curr Pharm Design https://doi.org/10.2174/1381612821666141211120604
12. Takebe N, Harris PJ, and Warren RQ, et al (2011) Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways Nat Rev Clin Oncol https://doi.org/10.1038/nrclinonc.2010.196
13. Chen CL, Hernandéz JDC, and Kumar DBU, et al (2023) Profiling of circulating tumor cells for screening of selective inhibitors of tumorinitiating stem-like cells Adv Sci (Weinheim, Baden-Wurttemberg, Germany) https://doi.org/10.1002/advs.202206812
14. Erdem M, Lee K, and Hardt M, et al (2024) MACC1 regulates LGR5 to promote cancer stem cell properties in colorectal cancer Cancers https://doi.org/10.3390/cancers16030604
15. Lu Y, Wang W, and Shi T, et al (2022) ScpEHD1/scp promotes the cancer stem cell (scpCSC/scp )-like traits of glioma cells via interacting with scpCD44/scp and suppressing scpCD44/scp degradation Environ Toxicol https://doi.org/10.1002/tox.23592
16. Lv J and Shim JS (2015) Existing drugs and their application in drug discovery targeting cancer stem cells Arch Pharm Res https://doi.org/10.1007/s12272-015-0628-1 PMID: 26152874
17. Ginestier C, Hur M, and Hur MH, et al (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome Cell Stem Cell https://doi.org/10.1016/j.stem.2007.08.014
18. Song J, Xie C, and Jiang L, et al (2018) Transcription factor AP-4 promotes tumorigenic capability and activates the Wnt/β-catenin pathway in hepatocellular carcinoma Theranostics https://doi.org/10.7150/thno.25194
19. Yang LX, Yang L, and Zhang CT, et al (2022) C1Q labels a highly aggressive macrophage-like leukemia population indicating extramedullary infiltration and relapse Blood https://doi.org/10.1182/blood.2022017046
20. McKeage MJ, Kotasek D, and Markman B, et al (2018) Phase IB trial of the anti-cancer stem cell DLL4-binding agent demcizumab with pemetrexed and carboplatin as first-line treatment of metastatic non-squamous NSCLC Target Oncol 13(1) 89–98 https://doi.org/10.1007/s11523-017-0543-0
21. Li C, Xie Q, and Ghosh S, et al (2025) SUV39H1 maintains cancer stem cell chromatin state and properties in glioblastoma JCI Insight https://doi.org/10.1172/jci.insight.186344
22. Fang Y, Xiao X, and Wang J, et al (2024) Cancer associated fibroblasts serve as an ovarian cancer stem cell niche through noncanonical Wnt5a signaling NPJ Parecis Oncol https://doi.org/10.1038/s41698-023-00495-5
23. Bushnell G, Sharma D, and Wilmot H, et al (2024) Natural killer cell regulation of breast cancer stem cells mediates metastatic dormancy Cancer Res https://doi.org/10.1158/0008-5472.CAN-24-0030 PMID: 39106452 PMCID: 11474167
24. Li Y, Wang W, and Xu D, et al (2024) PIWIL2/PDK1 axis promotes the progression of cervical epithelial lesions via metabolic reprogramming to maintain tumor-initiating cell stemness Adv Sci (Weinheim, Baden-Wurttemberg, Germany) https://doi.org/10.1002/advs.202410756
25. Xue M, Dong L, and Zhang H, et al (2024) METTL16 promotes liver cancer stem cell self-renewal via controlling ribosome biogenesis and mRNA translation J Hematol Oncol https://doi.org/10.1186/s13045-024-01526-9
26. Kimura T, Doolittle W, and Kruhlak M, et al (2024) Inhibition of MEK signaling attenuates cancer stem cell activity in anaplastic thyroid cancer Thyroid https://doi.org/10.1089/thy.2023.0521 PMCID: 10998707
27. Bonnet D and Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell Nat Med https://doi.org/10.1038/nm0797-730 PMID: 9212098
28. Croker AK, Goodale D, and Chu JE, et al (2009) High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability J Cell Mol Med https://doi.org/10.1111/j.1582-4934.2008.00455.x
29. Gupta PB, Gupta P, and Önder TT, et al (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening Cell https://doi.org/10.1016/j.cell.2009.06.034 PMID: 19682730 PMCID: 4892125
30. Ji Q, Hao X, and Zhang M, et al (2009) MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells PLoS One https://doi.org/10.1371/journal.pone.0006816
31. Kim JH, Chae M, and Kim WK, et al (2011) Salinomycin sensitizes cancer cells to the effects of doxorubicin and etoposide treatment by increasing DNA damage and reducing P21 protein Br J Pharmacol https://doi.org/10.1111/j.1476-5381.2010.01089.x
32. Mediratta K, El-Sahli S, and Marotel M, et al (2024) Targeting CD73 with flavonoids inhibits cancer stem cells and increases lymphocyte infiltration in a triple-negative breast cancer mouse model Front Immunol https://doi.org/10.3389/fimmu.2024.1366197 PMID: 38601156 PMCID: 11004431
33. Peyvandi S, Bulliard M, and Yilmaz A, et al (2024) Tumor-educated Gr1+CD11b+ cells drive breast cancer metastasis via OSM/IL-6/JAK-induced cancer cell plasticity J Clin Invest https://doi.org/10.1172/JCI166847 PMID: 38236642 PMCID: 10940099
34. Shu X, Xiong S, and Chen M, et al (2022) Palladin promotes cancer stem cell-like properties in lung cancer by activating Wnt/ scpβ-Catenin/scp signaling Cancer Med https://doi.org/10.1002/cam4.5192
35. Thayer SP, di Magliano MP, and Heiser PW, et al (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis Nature https://doi.org/10.1038/nature02009 PMID: 14520413 PMCID: 3688051
36. Zheng Q, Tang J, and Aicher A, et al (2023) Inhibiting NR5A2 targets stemness in pancreatic cancer by disrupting SOX2/MYC signaling and restoring chemosensitivity J Exp Clin Cancer Res CR https://doi.org/10.1186/s13046-023-02883-y
37. Sarkar R, Biswas S, and Ghosh R, et al (2024) Exosome-sheathed porous silica nanoparticle-mediated co-delivery of 3,3’-diindolylmethane and doxorubicin attenuates cancer stem cell-driven EMT in triple negative breast cancer J Nanobiotechnol https://doi.org/10.1186/s12951-024-02518-0
38. Chen W, Dong J, and Haiech J, et al (2016) Cancer stem cell quiescence and plasticity as major challenges in cancer therapy Stem Cells Int https://doi.org/10.1155/2016/1740936
39. Hanahan D and Weinberg RA (2000) The hallmarks of cancer Cell https://doi.org/10.1016/S0092-8674(00)81683-9
40. Xie X, Ganbold M, and Li J, et al (2024) Glioblastoma functional heterogeneity and enrichment of cancer stem cells with tumor recurrence Neuron https://doi.org/10.1016/j.neuron.2024.10.012 PMID: 39510072 PMCID: 11659040
41. Plaks V, Kong N, and Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell https://doi.org/10.1016/j.stem.2015.02.015 PMID: 25748930 PMCID: 4355577
42. Bray S and Bray SJ (2006) Notch signalling: a simple pathway becomes complex Nat Rev Mol Cell Biol https://doi.org/10.1038/nrm2009 PMID: 16921404
43. Zhou S, Schuetz JD, and Bunting KD, et al (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype Nat Med https://doi.org/10.1038/nm0901-1028
44. Aalam SMM, Nguyen LV, and Kannan N (2023) Clonal tracking in cancer and metastasis Cancer Metastasis Rev PMID: 37910295 PMCID: 11500829
45. Baniahmad A (2021) Tumor spheroids and organoids as preclinical model systems Med Genet PMID: 38835698 PMCID: 11006296
46. Blackburn JS and Langenau DM (2014) Zebrafish as a model to assess cancer heterogeneity, progression and relapse Dis Models Mech https://doi.org/10.1242/dmm.015842
47. Fuloria S, Subramaniyan V, and Gupta G, et al (2023) Detection of circulating tumor cells and epithelial progenitor cells: A comprehensive study J Environ Pathol Toxicol Oncol https://doi.org/10.1615/JEnvironPatholToxicolOncol.2022044456 PMID: 37017676
48. Siegel RL, Naishadham D, and Jemal A (2012) Cancer statistics, 2012 CA: A Cancer J Clin
49. Jin L, Hope KJ, and Hope KJ, et al (2006) Targeting of CD44 eradicates human acute myeloid leukemic stem cells Nat Med https://doi.org/10.1038/nm1483 PMID: 16998484 PMCID: 2754288
50. Zhu C and Stiehl T (2024) Modelling post-chemotherapy stem cell dynamics in the bone marrow niche of AML patients Sci Rep https://doi.org/10.1038/s41598-024-75429-7
51. Espinoza I and Miele L (2013) Notch inhibitors for cancer treatment Pharmacol Ther https://doi.org/10.1016/j.pharmthera.2013.02.003 PMID: 23458608 PMCID: 3732476
52. Castellón EA, Indo S, and Contreras HR (2022) Cancer stemness/epithelial–mesenchymal transition axis influences metastasis and castration resistance in prostate cancer: Potential therapeutic target Int J Mol Sci https://doi.org/10.3390/ijms232314917
53. Mather JP and Mather JP (2012) In vitro models Stem cells (Dayton, Ohio https://doi.org/10.1002/stem.774
54. Zhou L, Yu KHO, and Yu KH, et al (2021) Lineage tracing and single-cell analysis reveal proliferative Prom1+ tumour-propagating cells and their dynamic cellular transition during liver cancer progression Gut https://doi.org/10.1136/gutjnl-2021-324321
55. Shao Z, Wang H, and Ren H, et al (2023) The anticancer effect of napabucasin (BBI608), a natural naphthoquinone Molecules 28(15) 5678 https://doi.org/10.3390/molecules28155678 PMID: 37570646 PMCID: 10420168
56. Jonker DJ, Nott L, and Yoshino T, et al (2016). A randomized phase III study of napabucasin [BBI608] (NAPA) vs placebo (PBO) in patients (pts) with pretreated advanced colorectal cancer (ACRC): The CCTG/AGITG CO.23 trial Ann Oncol 27 vi150 https://doi.org/10.1093/annonc/mdw370.03





